肯尼迪病患者的临床与肌电图特点(附三例报告)
2015-06-15李靖李传芬胡怀强刘付红苏净高丽国曹秉振
李靖 李传芬 胡怀强 刘付红 苏净 高丽国 曹秉振
肯尼迪病患者的临床与肌电图特点(附三例报告)
李靖 李传芬 胡怀强 刘付红 苏净 高丽国 曹秉振
目的 探讨肯尼迪病(KD)的临床表现及肌电图特点。方法 总结3例经基因确诊的KD患者的临床资料,分析其临床、肌电图特点及实验室检查结果。结果 3例患者均表现为以肢体近端和延髓受累为主的下运动神经元损害,均可见上肢震颤,其中2例患者可见口周肌束震颤;3例患者血清肌酸激酶均升高(分别为1201、817、1247 U/L);3例患者肌电图均呈广泛的慢性神经源性损害,并存在感觉神经动作电位(SNAP)波幅降低、感觉神经传导速度(SCV)减慢等感觉神经病变的表现。3例患者雄激素受体基因外显子中CAG重复序列次数均>40(分别为48、51、52)。结论 KD的临床特点为缓慢进展的延髓和四肢肌肉萎缩无力,伴有内分泌或代谢异常;KD的肌电图表现为广泛神经源性损害,伴有感觉神经病变。
肯尼迪病;临床特点;肌电图
肯尼迪病(Kennedy disease,KD)是一种X-连锁隐性遗传病,其致病基因定位于X染色体长臂近侧端(Xq11-12),是临床少见病,其发病率为0.09/100000,患病率为1.6/100000[1]。现报道3例雄激素受体(androgen receptor,AR)基因检测阳性的KD患者,并分析其临床及电生理特点。
1 对象和方法
1.1 观察对象 2012-02-2013-05在作者医院神经内科就诊的KD患者3例,其中2例为门诊病例,1例为住院病例,均经AR基因检测明确诊断。
病例1:患者男,46岁,因“进行性四肢无力7年”于2012-02就诊。患者2005-02无明显诱因出现双下肢力弱,易疲劳,近端明显。在当地按腰椎间盘突出症治疗后症状无改善,呈缓慢进行性加重。3年后患者自觉双上肢力弱,提重物困难,四肢肌肉偶有“肉跳”,声音轻度嘶哑,无肢体麻木及疼痛,无饮水呛咳,体重无明显下降。既往体健。患者25岁结婚,育有1子,否认遗传疾病家族史。体格检查:发育正常,未见乳房女性化,外生殖器未见异常。神经系统查体:神志清楚,轻度构音障碍,可见舌肌纤颤,舌肌萎缩形成舌中沟;双上肢肩胛带肌欠饱满,双手大鱼际肌轻度萎缩;双上肢可见姿势性震颤,双上肢肌力Ⅴ-级,双下肢肌力Ⅳ级;共济运动稳准;深浅感觉正常;四肢腱反射未引出,病理反射(-)。
病例2:患者男,44岁。因“进行性四肢无力6年”于2012-11就诊。患者2006-10无明显诱因出现双下肢力弱,近端明显,呈进行性加重,2008-07出现双上肢力弱,双手震颤,在当地按周围神经病治疗未见明显好转,3年前出现双侧乳房肿胀、触痛,左侧明显,伴性功能减低。四肢肌肉偶有“肉跳”,声音轻度嘶哑,无饮水呛咳,6年来体质量减轻约5 kg。既往体健,22岁结婚,育有1子,否认遗传疾病家族史。体格检查:体型消瘦,双侧乳房肿大,左侧明显,有压痛,外生殖器未见异常。神经系统查体:神志清楚,轻度构音障碍;可见口周肌肉震颤,舌肌纤颤、萎缩及舌中沟,伸舌力弱;双手大、小鱼际肌、肱二头肌、双侧胫前肌均可见轻度萎缩,走路左右摇摆,鸭步步态,双上肢肌力Ⅳ级,下肢肌力Ⅲ级,共济运动稳准;深浅感觉正常;四肢腱反射未引出,病理反射(-)。
病例3:男性,44岁,因“进行性全身无力5年”于2013-04就诊。患者2008-05无明显诱因出现双下肢力弱,近端明显,2009-06出现双上肢力弱,双手震颤,并出现双侧乳房肿胀、触痛,左侧明显。四肢肌肉偶有“肉跳”,声音轻度变粗,无饮水呛咳,体质量无明显下降。既往体健,23岁结婚,育有1子、1女,一舅40余岁时有类似症状,未就诊,73岁死于肝癌。患者兄弟5人,一弟37岁,曾述腿痛,无其他症状,未就诊。体格检查:体型中等,双侧乳房肿大,左侧明显,有压痛,外生殖器未见异常。神经系统查体:神志清楚,轻度构音障碍,可见口周肌肉震颤,舌肌纤颤、萎缩,舌中沟明显(图1);双手大、小鱼际肌欠饱满;双上肢呈姿势性震颤,双上肢肌力Ⅴ-级,双下肢肌力Ⅳ级;四肢肌张力略低;共济运动稳准;深浅感觉正常;双下肢膝腱反射未引出,病理反射(-)。

图1 例3患者舌肌萎缩,舌中沟明显可见
1.2 方法 3例患者均进行血、尿常规,血生化(包括肝肾功能、血脂、心肌酶谱),性激素〔包括雌二醇、睾酮、黄体酮、泌乳素(PRL)、促黄体生成素、促卵细胞生成素(FSH)6项〕水平检查。应用丹麦产Keypoint电生理诊断仪对3例患者行常规针电极EMG和神经电图检测,EMG检查包括一侧咬肌、肱二头肌、第一骨间肌和对侧的T10椎旁肌、股四头肌、胫前肌6块肌肉。运动神经及感觉神经检查均包括双侧正中神经、尺神经、腓总神经、胫神经8条神经,同时检测正中神经的F-波和胫神经H-反射。
在3例患者知情同意下分别采集静脉血4 mL提取受检者基因组DNA,利用PCR方法扩增AR基因第1外显子,经电泳片段克隆测序。
2 结果
3例患者肌酸激酶(CK)水平均明显升高(分别为1201、817、1247 U/L,正常值参考值范围:26~200 U/L),例1和例3患者三酰甘油(TG)轻度升高(分别为3.49、3.19 mmol/L,正常值参考值范围:0.50~1.95 mmol/L),例2患者睾酮水平升高(846.30 ng/dL,正常值参考值范围:175~781 ng/dL),例3患者血糖轻度升高(6.11 mmol/L),PRL(379.9,正常值参考值范围:86~324 uIU/mL)、FSH(14.02 IU/L,正常值参考值范围:1.5~12.4 IU/L)增高,3例患者余生化指标均正常(表1)。例2患者行腰椎穿刺检查,脑脊液常规、生化、寡克隆电泳分析均正常。
3例患者所检测的肌肉在静息状态下均可见自发电位,运动单位电位时限增宽、波幅增高,大力收缩呈单纯相,提示广泛神经源性损害。运动神经传导速度(MCV)均正常,复合肌肉动作电位(compound muscle action potentials,CMAP)波幅正常或轻度减低;3例患者共检测24条感觉神经,其中3条(例1患者2条、例2患者1条)感觉神经传导速度(SCV)减慢,16条(例1、例3患者各6条,例2患者4条)感觉神经动作电位(sensory nerve action potential,SNAP)波幅降低,2条未引出波形,例1患者H-反射未引出,例2患者H-反射波幅低。
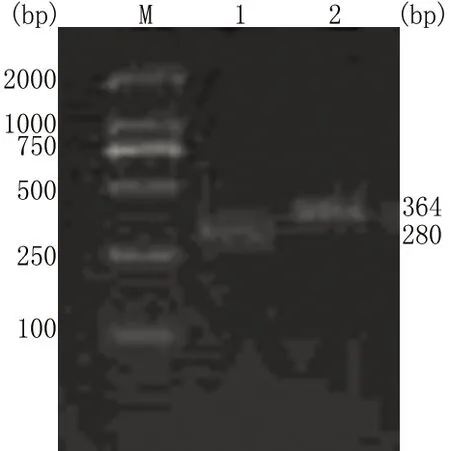
M:DL 2000 DNA Marker;1:健康对照者;2:KD患者
图3 例1患者AR基因PCR产物电泳图

图2 例1患者的PCR产物AR基因第1外显子基因测序结果:CAG重复序列次数为48次
3例患者的AR受体第1外显子编码的CAG三核苷酸重复重复序列次数均>40,分别为48、51、52(图2、3)。
3 讨论
KD是一种起病于30~50岁之间X-连锁隐性遗传,累及下运动神经元脊髓延髓型肌萎缩,1968年由Kennedy等[2]首先进行了系统的临床和病理研究。1991年La Spada等[3]报道在健康人群中雄激素受体第一外显子编码的CAG三核苷酸重复17~35次,而KD患者CAG三核苷酸重复超过40次,并认为CAG三核苷酸重复扩增是其主要致病基因,该基因的重复扩增选择性导致脊髓和延髓运动神经元变性,上运动神经元不受累及,病理可见脊髓前角细胞和脑干下部运动神经核团的神经元减少。由于该病神经肌肉损害的临床症状与肌萎缩侧索硬化(ALS)、脊髓性肌萎缩、遗传性感觉运动神经病、肌营养不良等疾病有交叉、相似之处,极易引起误诊。临床的早期确诊有助于减少经济负担,增强患者的生活信念,以便提供必要遗传咨询支持。
KD的确诊需要依靠基因检测,本文3例患者AR基因外显子中CAG重复序列次数均>40,确诊为KD。作者通过对基因确诊的3例KD患者的临床及电生理特点分析,进一步探讨KD的独特临床特征。
此3例KD患者均临床起病缓慢,逐渐进展,以双下肢无力起病,呈对称性,并逐渐累及上肢,且均以四肢近端无力为主,伴有肌肉萎缩及上肢姿势性震颤。3例患者均有舌肌萎缩,舌中线附近肌肉萎缩显著,舌中沟明显可见。文献报道约95%以上的KD患者具有面部尤其是口周肌束震颤,这在其他遗传或获得性神经系统变性病中少见,是该病显著的临床特征[4]。本组3例患者中2例可见到明显的口周肌束震颤。此外,本组中2例患者出现了男性乳房女性化,3例患者CK水平均明显增高,例1和例3患者TG轻度升高,例2患者睾酮升高,例3患者血糖轻度升高,PRL、FSH升高,说明本病患者临床也可表现出男性乳房发育、性功能减退以及血脂、血糖水平异常等内分泌或代谢异常表现,这与文献[5]报道一致,也是较其他运动神经元病的不同之处。
EMG呈神经源性损害,感觉神经动作电位缺失或波幅降低也是KD的电生理学特点,这也是与运动神经元病鉴别的重要方面。肌电图检查显示3例患者存在广泛神经源性损害,累及咬肌、上下肢远近端肌肉和胸段椎旁肌,提示脑神经运动核及脊髓前角细胞损害。神经电生理结果显示运动神经传导速度正常,CMAP波幅正常或轻度减低,减低程度与肌萎缩程度相当;3例患者尽管体格检查均无感觉异常,但是感觉神经传导检测均出现全部或部分异常,且以SNAP波幅的减低为主,与文献[6]报道一致。KD患者肌电图和运动神经传导的表现均类似于ALS的电生理改变,但其感觉神经元也可发生变性,导致感觉系统受累,而ALS患者并不累及感觉神经,两者可藉此鉴别。
综上可见,随着临床对该病的深入研究,加深对临床症状的了解,尤其对其电生理特点的进一步认识,可提高本病的早期诊断率。对于缺乏基因诊断的地区,通过详细的神经系统查体和神经电生理检查有助于尽早拟诊此病。
[1]Guidetti D, Sabadini R, Ferlini A,et al. Epidemiological survey of X-linked bulbar and spinal muscular atrophy,or Kennedy disease,in the province of Reggio Emilia,Italy[J].Eur J Epidemiol,2001,17:587-591.
[2]Kennedy WR,Alter M,Sung JH.Progressive proximal spinal and Bulbar muscular atrophy of late onset. A sex-linked recessive trait[J].Neurology,1968,18:671-680.
[3]La Spada AR,Wilson EM,Lubahn DB,et al.Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy[J].Nature,1991,352:77-79.
[4]Paparounas K. Kennedy disease: insights and questions[J].Arch Neurol,2004,61:603.
[5]Greenland KJ,Zajac JD.Kennedy’s disease: pathogenesis and clinical approaches[J].Intern Med J, 2004, 34:279-286.
[6]鲁明,张俊,郑菊阳,等.12例肯尼迪患者肌电图和神经电图特点[J].中国神经免疫学和神经病学杂志,2008,15:187-189.
(本文编辑:邹晨双)
The clinical and electromyogram features of Kennedy disease (a report of three cases)
LIJing*,LIChuanfen,HUHuaiqiang,LIUFuhong,SUJing,GAOLiguo,CAOBingzhen.
*DepartmentofNeurology,JinanMilitaryGeneralHospital,JinanShandong250031,China
LI Jing, Email:love.jingtong@163.com
Objective To summarize the clinical and electromyogram features of Kennedy disease (KD), to further understand KD. Methods We analyzed the clinical manifestation, electromyogram features, and laboratory examinations of three KD patients whose diagnosis were defined by gene analysis. Results All of the cases were characterized by lower motor neuron damage predominantly in the proximal and bulbar muscle, accompanied by upper limb tremor and high levels of creatine kinase (1201 U/L,817 U/L,1247 U/L,respectively). There were obvious perioral muscle tremors in two cases. EMG detected a widespread neuronal damage, decreased amplitude of SNAP, and SCV in all three cases. Three CAG repeat numbers in AR gene were all above 40(48,51,52,respectively). Conclusions KD is characterized by slow progress of bulbar and limb muscle weakness and atrophy, associated with endocrine or metabolic disturbance. KD showed neurogenic damage, accompanied by sensory neuropathy on electromyography.
Kennedy disease;clinical features;electromyogram
10.3969/j.issn.1006-2963.2015.02.003
250031济南军区总医院神经内科
李靖,Email:love.jingtong@163.com
R746.9
A
1006-2963 (2015)02-0086-04
2014-01-14)
