动态亚胺化学的分子设计及其动态性质研究
2024-01-14刘小红刘家铭秦静静梁利岩
刘小红,刘家铭,秦静静,梁利岩
(1.中国科学院广州化学研究所,广东 广州 510650;2.中国科学院大学,北京 100049;3.中国科学院新型特种精细化学品工程实验室,广东 广州 510650;4.国科广化韶关新材料研究院,广东 南雄 512499;5.国科广化(南雄)新材料研究院有限公司,广东 南雄 512499)
随着动态化学的发展,依赖于动态共价键的动态共价交联自适应性网络被提出[1-3];并且该类型的聚合物网络可同时具备热固性树脂和热塑性塑料的优势,通过动态交联之间的可逆反应使得聚合物网络中的分子链重新排列,进而表现出再加工、自愈合或回收性能[4-5]。
动态共价键(dynamic covalent bonds,DCBs)作为一类能够在一定外界刺激(包括热、光、pH 等)下可逆断裂和重组的化学键,因其具有独特的响应性而受到很多关注[6-9]。然而,在动态共价键的化学宝库中,由醛或酮与伯胺经脱水缩合反应生成的亚胺键是最为经典的化学键之一。它于1864 年被德国化学家Hugo Schiff 首次发现。亚胺键可进行三类动态反应,包括氨基转移反应、亚胺复分解反应和亚胺缩合/水解反应。因此,亚胺键也被认为是一种最具有代表性的动态可逆键,这使它在制备动态共价聚合物中被广泛采用[10-13]。
因此,在本文中,通过分子设计,合成相关亚胺小分子单体,并对亚胺键的三类动态特性进行了相关验证。首先,将两类不同的亚胺小分子模型化合物进行混合,采用核磁氢谱和GC-MS 分析方法对亚胺键的复分解反应机制进行了分析。其次,亚胺键的氨基转移反应也通过核磁表征而在亚胺小分子单体和另一分子伯胺化合物中得到了验证。此外,亚胺键的水解机制也被详细证明。
1 实验
1.1 试剂
苄胺(AR,≥99%),4-羟基苯甲醛(AR,≥99%)和4-甲氧基苯胺(AR,≥97%)上海麦克林生化科技有限公司;苯甲醛(AR,≥99%),4-氨基苯酚(AR,≥99%),苯胺(AR,≥99%),4-硝基苯甲醛(AR,≥97%),阿拉丁化学试剂上海公司;无水乙醇和石油醚,AR,天津大茂化学试剂厂。
1.2 相关亚胺小分子单体的合成
1.2.1 (E)-N,1-二苯甲基苯胺(1)
首先,将苯甲醛(4.24 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶中,随后将苯胺(3.72 g,40 mmol)滴入混合液内,观察到混合液逐渐变黄。将反应液升温至60℃进行搅拌12 h 后冷却至室温,此时将反应液置于50~60℃的旋转蒸发仪中减压除去有机溶剂得到大量微黄色固体粉末粗品。用少量无水乙醇溶解粗品,然后将其滴入到大量去离子水中,发现大量乳白色晶体析出。最后,将收集的乳白色晶体置于真空70℃干燥24 h 至恒重,得到(E)-N,1-二苯甲基苯胺(1)(6.06 g,产率83.67%)。具体的合成路线及结构表征如图1 和图2 所示。
图1 单体1 的合成路线
1H-NMR(400 MHz,CDCl3,δ,ppm): 8.46(s,1H,N=C-H),7.94-7.91(d,2H,Ar-H),7.50-7.45(m,3H,Ar-H),7.42-7.38(d,2H,Ar-H),7.24-7.20(m,3H,Ar-H)
13C-NMR(400 MHz,CDCl3,δ,ppm): 160.51,152.09,136.20,131.44,129.19,128.84,128.82,125.98,120.90
1.2.2 (E)-N-(4-甲氧基亚苄基)-1-(4-硝基)苯胺(2)
将4-硝基苯甲醛(6.05 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶内,然后将4-甲氧基苯胺(4.93 g,40 mmol)滴入其中,观察到有荧黄色沉淀生成。将反应液升温至60℃继续磁力搅拌12 h 后冷却至室温,所得固体物质过滤后用无水乙醇洗涤数次,最后,将固体物质于真空70℃干燥24 h 至恒重,得到黄色粉末物质(E)-N-(4-甲氧基亚苄基)-1-(4-硝基)苯胺(2)(9.85 g,产率96.17%)。具体的合成路线及结构表征如图3 和图4 所示。
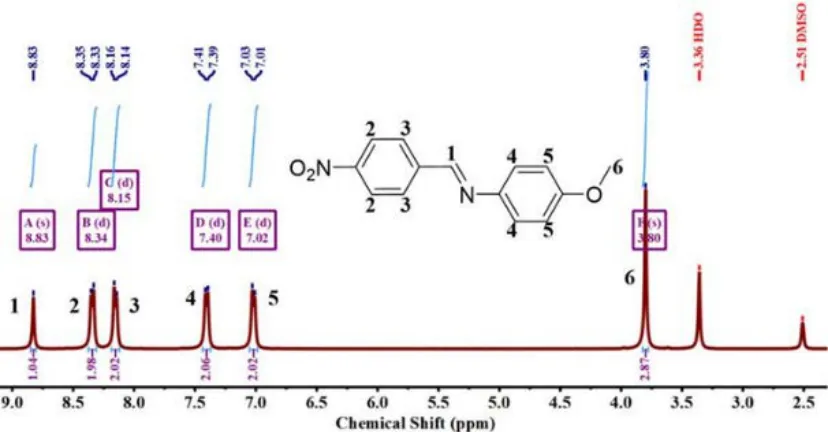
图4 单体2 的1H-NMR 谱图
1H-NMR(400 MHz,DMSO-d6,δ,ppm): 8.83(s,1H,N=C-H),8.34(d,2H,Ar-H),8.15(d,2H,Ar-H),7.40(d,2H,Ar-H),7.02(d,2H,Ar-H),3.80(s,3H,OCH3)
13C-NMR(400 MHz,DMSO-d6,δ,ppm): 164.26,158.11,149.65,144.18,140.16,129.04,124.48,121.72,114.07,55.34
1.2.3 (E)-1-(4-硝基亚苄基)-N-苯胺(3)
将4-硝基苯甲醛(6.05 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶内,然后将苯胺(3.72 g,40 mmol)滴入其中,观察到有微黄色沉淀生成。将反应液升温至60℃继续磁力搅拌12 h 后冷却至室温,所得固体物质过滤后用无水乙醇洗涤数次,最后,将固体物质于真空70℃干燥24 h 至恒重,得到黄色粉末物质(3)(8.63 g,产率95.43%)。具体的合成路线及结构表征如图5 和图6 所示。

图5 单体3 的合成路线

图6 单体3 的1H-NMR 谱图
1H-NMR(400 MHz,DMSO-d6,δ,ppm): 8.81(s,1H,N=C-H),8.36(d,2H,Ar-H),8.19(d,2H,Ar-H),7.46(d,2H,Ar-H),7.35(d,2H,Ar-H),7.32(m,1H,Ar-H)
13C-NMR(400 MHz,DMSO-d6,δ,ppm): 160.61,151.51,149.06,139.89,129.36,129.04,126.31,124.48,121.05
1.2.4 (E)-N-(4-甲氧基亚苄基)苯胺(4)
将苯甲醛(4.24 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶内,然后将4-甲氧基苯胺(4.93 g,40 mmol)滴入其中,观察到有微黄色沉淀生成。此时,将反应液升温至60℃继续反应12 h 后冷却至室温,所得固体物质过滤后用无水乙醇洗涤数次,最后,将固体物质于真空70℃干燥24 h 至恒重,得到黄色粉末物质(E)-N-(4-甲氧基亚苄基)苯胺(4)(8.23 g,产率98.28%)。具体的合成路线及结构表征如图7和图8 所示。
图7 单体4 的合成路线

图8 单体4 的1H-NMR 谱图
1H-NMR(400 MHz,DMSO-d6,δ,ppm): 8.65(s,1H,N=C-H),7.94-7.90(d,2H,Ar-H),7.55-7.50(m,3H,Ar-H),7.33-7.29(d,2H,Ar-H),7.01-6.97(d,2H,Ar-H),3.79(s,3H,-OCH3)
13C-NMR(400 MHz,DMSO-d6,δ,ppm): 160.64,158.16,145.20,135.83,130.40,128.85,128.74,121.75,114.07,55.31
1.2.5 (E)-N-苯亚甲基-1-苯基甲胺(5)
首先,将苯甲醛(4.24 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶内,随后将苄胺(4.28 g,40 mmol)滴入混合液中,观察到混合液逐渐变黄。将反应液升温至60℃反应12 h 后,再将其置于50~60℃的旋转蒸发仪中减压旋蒸除去有机溶剂,直到出现大量微黄色固体粉末粗品。用少量无水乙醇溶解粗品,然后将其滴入到大量去离子水中,发现大量乳白色晶体析出。最后,将收集的灰白色晶体置于真空70℃干燥24 h 至恒重,得到(E)-N-苯亚甲基-1-苯基甲胺(5)(6.80 g,产率87.24%)。具体的合成路线及结构表征如图9 和图10 所示。
图9 单体5 的合成路线
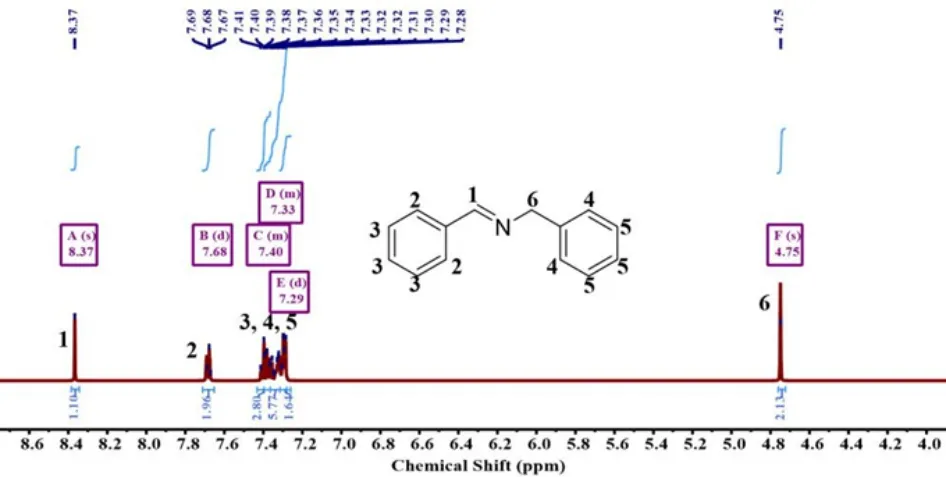
图10 单体5 的1H-NMR 谱图
1H-NMR(400 MHz,DMSO-d6,δ,ppm): 8.47(s,1H,N=C-H),7.69-7.67(d,2H,Ar-H),7.41-7.35(m,3H,Ar-H),7.34-7.31(m,2H,Ar-H),7.30-7.28(m,3H,Ar-H),4.75(s,2H,-NCH2-)
13C-NMR(400 MHz,DMSO-d6,δ,ppm): 160.20,138.76,135.73,129.74,128.70,128.53,128.27,128.05,127.28,62.38
1.2.6 (E)-4-((4-羟基亚苄基)氨基)苯酚(6)
首先,将对羟基苯甲醛(4.88 g,40 mmol)溶于100 mL 无水乙醇并加入到一个带回流-搅拌装置的250 mL 三颈烧瓶中,然后将对氨基苯酚(4.36 g,40 mmol)滴入到混合液内,观察到混合液逐渐变浅红棕色。将混合物加热并在60℃反应24 h 后,将其置于50~60℃的旋转蒸发仪中除去有机溶剂,直至出现大量微红色固体粉末粗品。用少量无水乙醇溶解粗品,随后加入冰石油醚,发现大量微红色固体物质析出,过滤除去溶剂,并通过少量多次方法用无水乙醇洗涤数次。最后,将收集的固体置于真空70℃干燥24 h 至恒重,得到纯物质(6)(7.83 g,产率91.84%)。具体的合成路线及结构表征如图11 和图12 所示。
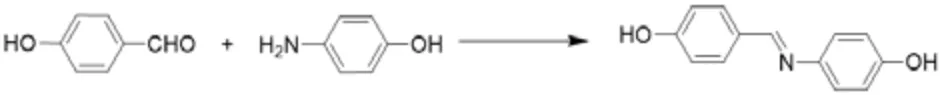
图11 单体6 的合成路线
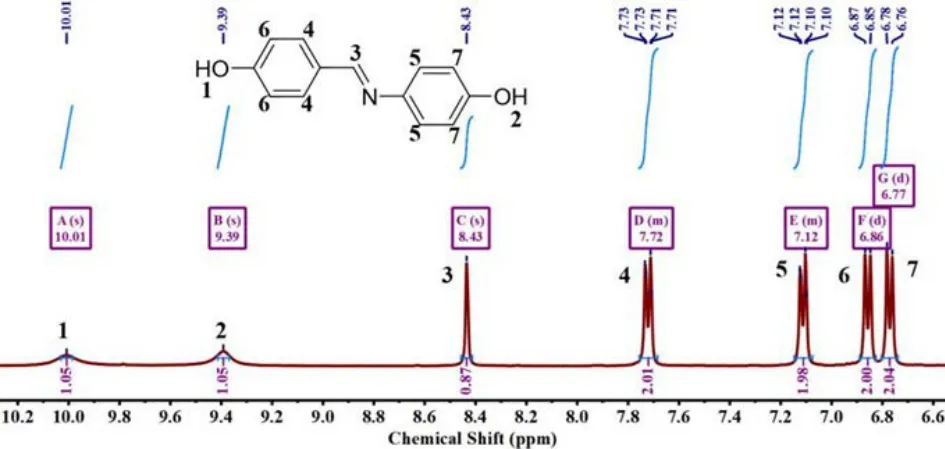
图12 单体6 的1H-NMR 谱图
1H-NMR(400 MHz,DMSO-d6,δ,ppm): 10.01(s,1H,Ar-OH),9.39(s,1H,Ar-OH),8.43(s,1H,N=C-H),7.73-7.71(d,2H,Ar-H),7.12-7.10(d,2H,Ar-H),6.87-6.85(d,2H,Ar-H),6.78-6.76(d,2H,Ar-H)
13C-NMR(400 MHz,DMSO-d6,δ,ppm): 160.78,157.01,156.85,141.62,131.40,127.36,123.89,117.23,116.62
1.3 亚胺小分子的亚胺复分解反应机理
为了验证亚胺的复分解反应,将适量亚胺小分子单体(1)置于含有氘代二甲亚砜的核磁样品管中,待收集到(1)的核磁氢谱后,取适量亚胺小分子单体(2)加入其中再次进行核磁氢谱表征;随后在核磁共振谱仪中选取混合样品不同反应时间(10 min、30 min、1 h 和3 h)的核磁氢谱。
另外,还采用GC-MS 光谱分析进一步验证亚胺复分解反应。具体的,将适量亚胺小分子单体(6)溶于含有少量无水乙醇的样品管中,随后加入适量(1)混匀,通过监测化学物质的保留时间和质谱来分析混合样品的反应情况。
1.4 亚胺小分子的氨基转移反应机理
为了验证亚胺的氨基转移(转胺交换)反应,将适量亚胺小分子单体(1)置于含有氘代二甲亚砜的核磁样品管中,待收集到(1)的核磁氢谱后,取适量苄胺加入其中再次进行核磁氢谱表征;随后在核磁共振谱仪中选取混合样品不同反应时间(10 min、30 min 和1 h)的核磁氢谱。
1.5 亚胺小分子的亚胺水解反应机理
同样通过核磁氢谱法对亚胺的水解反应进行了验证,首先取适量苯甲醛溶于含有氘代二甲亚砜中进行核磁氢谱表征,另取核磁管采用相同方法表征苯胺的核磁氢谱。随后,将适量苯甲醛和苯胺同时加入含有氘代二甲亚砜中进行核磁氢谱表征,待收集谱图后,取适量新配置的0.1 mol/L 的盐酸水溶液(氘代二甲亚砜和水的比例为8∶2)加入其中以进行水解反应,并收集其混合物的核磁氢谱。另外,另取含有1 mL 的0.1 mol/L 盐酸水溶液(氘代二甲亚砜和水的比例为8∶2)的核磁管,将适量苯甲醛溶于其中,再取适量苯胺加入混合液中,待混合液充分混匀后进行核磁氢谱表征。
1.6 测试与表征
1H-NMR 和13C NMR 是在室温下采用BRUKER AVANCE 400 光谱仪记录的,其中,以四甲基硅烷(TMS)为内标物的CDCl3或DMSO-d6为溶剂溶解样品。
GC-MS 分析采用7890B-5977A 气相色谱-质谱仪(GC-MS,美国安捷伦)进行测试。
2 结果与讨论
2.1 亚胺复分解反应机理验证
为了研究亚胺复分解反应的交换机理,合成相关亚胺小分子单体模型来验证亚胺键的复分解反应,见图13。

图13 亚胺键的复分解反应机理图
将制备的亚胺小分子单体1 与2 以特定的化学计量比置于含有DMSO-d6的核磁样品管中,通1HNMR 谱图中的氢谱变化来测定两种亚胺小分子单体的化学反应过程。正如图14 所示,将单体1 与2在核磁管中混合后立即进行测试,10 min 后,化学位移在8.65 ppm 处便已出现新的亚胺特征峰,通过与纯单体3 和纯单体4 的核磁氢谱进行对比,确认8.65 ppm 处的新峰归属于单体4 的亚胺特征峰。并且,化学位移在 7.48 ppm 和7.0 ppm 处也出现新的特征峰,这两类峰也归属于单体4 的特征峰。另外,在8.38 ppm 和8.18 ppm 处也出现了新的特征峰,经确认该两类峰归属于单体3 的相关氢质子峰。上述结果说明亚胺小分子单体1 与单体2 在室温下发生了动态交换反应。
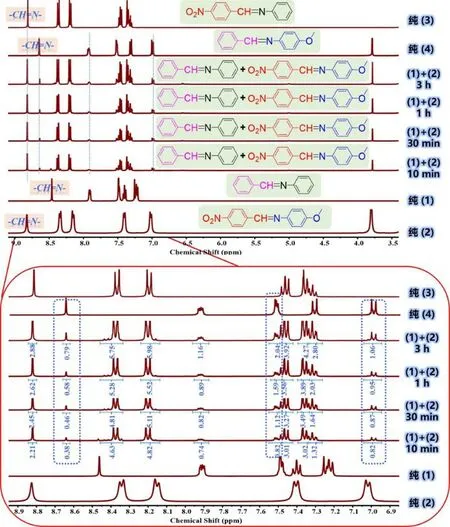
图14 亚胺小分子1 和2 在不同反应时间内的核磁氢谱图
在理想状态下,根据动态亚胺键的复分解反应机制(图13),亚胺小分子单体1 与单体2 的动态交换反应产物有四种,分别为:原料单体1、2 和含新的亚胺键的产物3 和4。然而,图中可以明显检测到新单体3 和4 的相关特征峰,并且特征峰强度在明显增强,而原料1 和2 的相关峰却明显减弱。正如图15 所示,随着反应的进行(10 min、30 min、1 h 和3 h),混合物中单体1 中8.45 ppm 处的亚胺特征峰以及亚胺小分子单体2 在化学位移为8.35 ppm 和8.15 ppm 处的相关特征峰都在不断减弱;而所有新出现的单体3 和4 的特征峰强度却在明显增大。这可能是因为混合物的动态交换反应在持续向正方向进行,并且随着反应时间的不断延长,正向反应产物单体3 和4 的浓度不断增大,而原料1 和2 的浓度不断减小,因而原料单体的峰会减弱甚至被产物3 和4 的强峰掩盖。
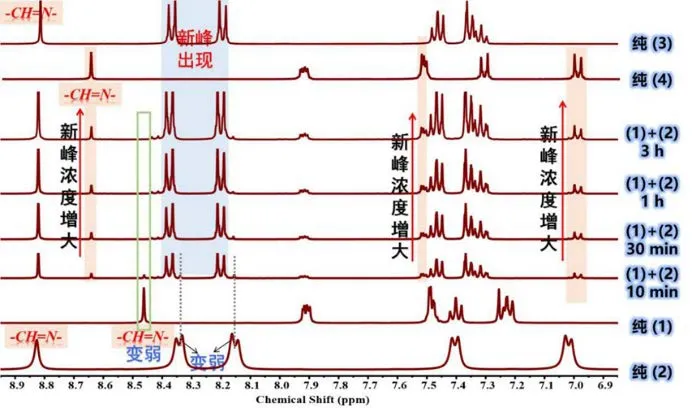
图15 亚胺小分子1 和2 在不同反应时间内的核磁氢谱图
此外,还通过GC-MS 分析来进一步确定亚胺键的复分解反应机制。具体的,通过将亚胺小分子单体1 和6 以一定当量比混合反应,通过GC-MS监测混合物的动态反应过程来确定亚胺键的交换反应机制。
如图16a 所示,GC-MS 监测到一定强度的三种物质特征峰,其保留时间分别为16.17、20.05 和23.31 min,这表明两种反应单体之间发生了化学反应。进一步的,通过质谱进行结构鉴定(表1),可确定不同流出时间所对应的不同化合物。其中,流出时间为16.17 和23.31 min 的色谱峰分别对应于原料单体1 和单体2。而保留时间为20.05 min 出现了一个新的强峰,结合质谱分析,对该峰的物质分子量以及所对应的碎片峰进行对比,最终确认了新出现的峰恰好归属于亚胺复分解反应(图16b)后的产物。
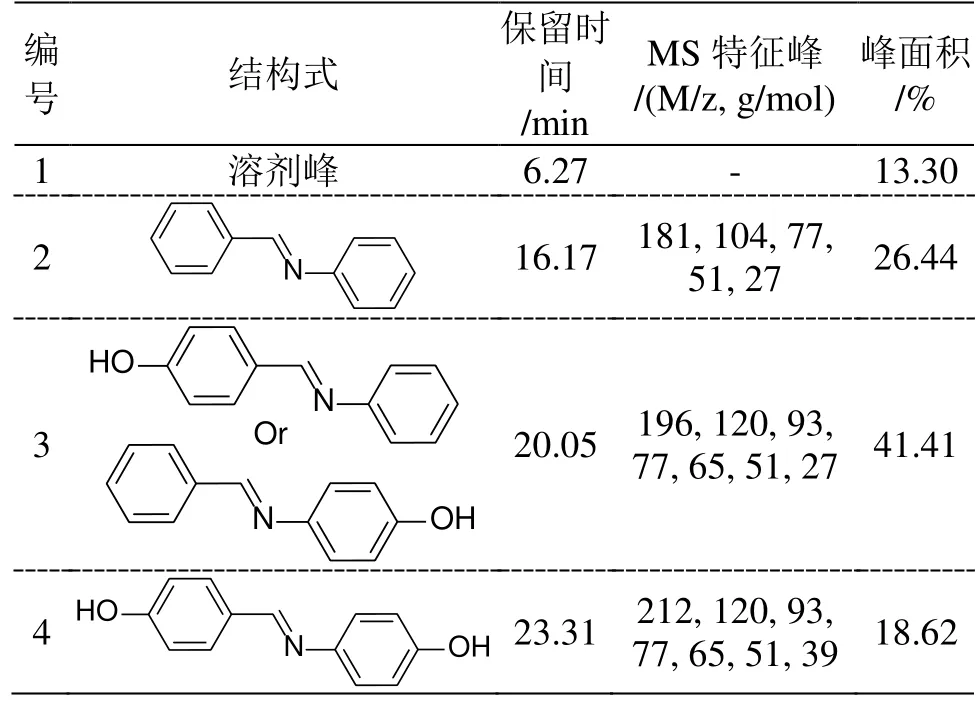
表1 GC-MS 谱图峰的分布
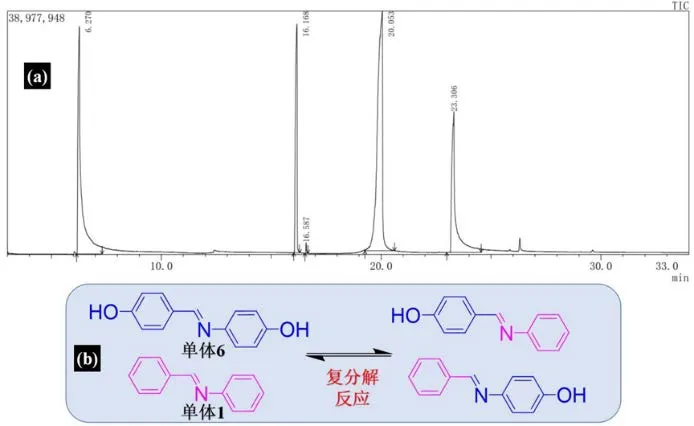
图16 亚胺小分子单体1 和亚胺小分子6 在室温下混合后a.气相色谱图;b.单体1 与单体6 亚胺复分解反应
因此,通过以上的分析,一定程度验证了动态亚胺键的复分解反应机理,并且该交换机理可进一步指导动态亚胺聚合物网络的合成与制备,以及验证其所带来的各项动态特性,如可自愈性[14]、可焊接性[15]、重塑再加工[16]等。
2.3 亚胺的氨基转移反应机理验证
在上一小节中,通过核磁氢谱和GC-MS 分析对亚胺键的复分解反应进行了验证;而为了阐明亚胺键的动态氨基转移反应机制,也通过图17 所示的亚胺小分子单体模型对其进行了研究。将成功制备的亚胺小分子模型单体1 和苄胺以特定的化学计量比置于含有DMSO-d6的核磁样品管中,通过核磁氢谱变化研究了亚胺键的氨基转移反应过程。如图18a 所示,单体1 与苄胺的混合10 min 后,1HNMR 能监测到一定强度的产物峰(在8.49 ppm、7.95 ppm、7.54 ppm、6.75 ppm 以及4.62 ppm 处都出现了新的氢质子峰),这表明混合物进行了相关的化学反应。图18a 中分别显示了不同反应时间的混合物核磁氢谱图与原料单体、纯单体5 以及苯胺的核磁氢谱的对比,经比对确认8.49 ppm、7.95 ppm、7.54 ppm 以及4.62 ppm 处的新峰归属于单体5 的特征峰,其中,8.49 ppm 为亚胺特征峰。另外,在6.75 ppm 及4.62 ppm 处的化学位移为苯胺的氢质子特征峰。上述结果说明亚胺小分子单体5 与苄胺在室温下发生了化学反应,其反应机制正如图17所示,即单体1 与苄胺在室温无催化剂下通过亚胺氨基转移反应生成了新的含亚胺键的产物5 和苯胺单体。
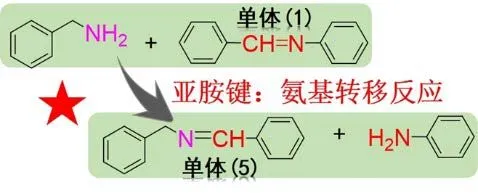
图17 亚胺键的氨基转移反应机理图

图18 亚胺单体1 和纯苄胺在不同反应时间内的(a)核磁氢谱图和(b)局部放大谱图
对核磁谱图的进一步分析后,如图18b 所示,可以明显观察到原料1 的相关特征信号(8.45 ppm)在反应过程中不断降低;然而产物5 和苯胺的相关特征峰信号却明显增强。这说明氨基转移反应在不断向产物方向进行,并且随着反应时间的延长,原料会不断消耗,产物持续生成,直至达到动态平衡状态[17]。
2.4 亚胺水解反应机理验证
众所周知,亚胺键是由羰基化合物醛或酮与伯胺经脱水缩合脱水而形成的[18],该反应在室温下无需催化剂即可进行。在前面的研究中,已对亚胺键的复分解反应和氨基转移反应进行了分析,然而,水解反应是动态亚胺键的又一明显特征。在酸和H2O 的催化下,亚胺键可发生水解反应并重新生成醛基和氨基化合物,如图19 所示。

图19 亚胺键的水解反应机理图
与前期的研究一致,同样采用了核磁表征方式对亚胺键的水解反应进行了探索。首先,如图20 所示,将苯甲醛和苯胺的混合物核磁氢谱与纯苯甲醛、苯胺、纯单体1 的核磁氢谱进行对比,可以发现,混合物核磁谱图(蓝色谱图)在8.45 ppm 处已开始出现新的亚胺特征峰。此外,在7.2 ppm 及7.0 ppm 处也开始出现新的质子峰。这说明苯甲醛与苯胺在室温下无需催化剂即可进行脱水缩合形成亚胺键。而当在苯甲醛与苯胺的混合物中加入0.1 mol/L 的盐酸水溶液(氘代二甲亚砜和水的比例为8∶2)时,8.45 ppm 处的亚胺峰却消失不见(红色谱图),由此表明酸催化下醛基与氨基无法进行缩合反应,因此亚胺键也无法形成。

图20 相关亚胺小分子的核磁氢谱图
此外,除了纯单体1 的核磁谱图外,还在物质1 中加入了0.1 mol/L 的盐酸水溶液(氘代二甲亚砜和水的比例为8∶2)进行氢谱检测用以确认亚胺键的水解反应。如图20 中的绿色谱图所示,加入酸后物质1 的混合物氢谱新增了三处质子峰,其化学位移分别为9.75 ppm、7.5 ppm 和4.65 ppm;然而位于8.45 ppm 处的特征峰却已消失不见。在经过与纯单体1,纯苯甲醛以及纯苯胺的核磁氢谱图对比后,可以发现9.75 ppm、7.5 ppm 处新增特征峰分别归属于苯甲醛的醛基和苯环的质子峰;而化学位移为4.65 ppm 的新增质子峰为苯胺的氨基特征峰;同时,在8.45 ppm 处消失的质子峰则为单体1 的亚胺特征峰。
基于以上结果分析,可以发现,亚胺键在室温下无需催化剂即可由醛基和氨基经缩合反应形成;同时在酸性条件下它又极易发生水解反应而重新生成原来的醛基和氨基单体。因此,得益于亚胺键简单的制备过程和拥有独特的水解反应机制,基于亚胺键制备的动态自适应聚合物材料在一定程度上可以进行降解回收,一方面,可以缓解不可降解的聚合物材料所带来的环境压力;另一方面,也为高价值原料的可回收提供可能性。
3 结论
如图21 在本工作中,通过分子设计,合成了不同的亚胺小分子模型化合物。主要通过在线核磁氢谱和GC-MS 分析手段对亚胺键的复分解反应、氨基转移反应以及亚胺水解反应进行了确认,这为将其引入到高分子材料所引发的动态交换特性的应用中提供了理论基础。结果表明,不同亚胺小分子之间可在室温无催化剂条件下发生亚胺键的复分解反应;其次,亚胺键的氨基转移反应也可在无需催化剂引发下通过亚胺小分子单体和另一分子苄胺中体现;最后,在酸性水溶液中,亚胺键可在室温下发生水解反应,并将其水解成原先含醛基和氨基的小分子单体。
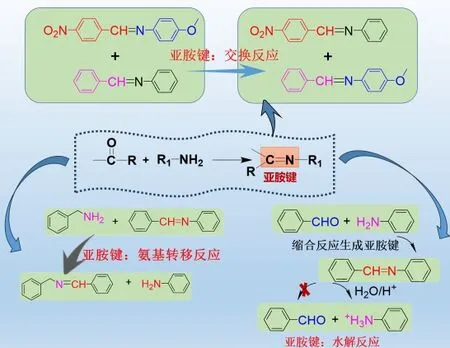
图21 动态亚胺化学的相关小分子设计示意图
