两种酪胺磁性分子印迹聚合物的 制备及比较
2018-09-22于芳芳师晓漫于欣欣
张 灿,于芳芳,师晓漫,于欣欣
(江苏大学食品与生物工程学院,江苏镇江 212013)
酪胺(Tyramine,TM)是酪氨酸在氨基酸脱羧酶的作用下产生的一种含氮低分子生物胺[1]。生物胺广泛存在于各种食品中,如酒类、调味品(醋和酱油)中。酪胺浓度过高时不仅会影响食品的风味,而且还导致心律加快,引发偏头疼等问题[2],此外含量的高低也被用于判断食品的新鲜程度。目前常用的检测生物胺的方法主要有薄层色谱[3]、高效液相色谱[4]等方法。但这些方法成本较高,在检测前都需要进行复杂的预处理,效果不太理想。然而,由于食品基质的复杂性和分析物的低浓度,分离和富集酪胺是样品预处理步骤的关键。
分子印迹聚合物(Molecularly imprinted polymers,MIPs)是对模板分子具有高度选择性的人造多孔材料[5]。因为其化学稳定性以及成本低等优点,已被广泛应用于固相萃取和传感器等诸多领域[6-7],但传统的分子印迹聚合物也存在模板分子泄漏,传质速度慢,分离过程复杂等缺点[8]。
本文采用表面分子印迹和磁性纳米技术相结合的方法,选择羧甲基葡聚糖包被的磁性纳米粒子(CM-Fe3O4)作为载体,酪胺作为模板分子,成功制备了酪胺的磁性分子印迹聚合物(dex-MMIPs),对酪胺具有良好的吸附能力。同时以硅烷偶联剂修饰的磁性纳米粒子(Fe3O4@SiO2-MPS)为载体合成了MPS-MMIPs,通过吸附实验比较这两种印迹聚合物的性能。
1 材料与方法
1.1 材料与仪器
酪胺(Tyramine,TM)、烟酰胺(Nicotinamide,VPP)(如图1所示)、六水合氯化铁、四水合氯化亚铁和葡聚糖(MW 20000) 购自国药集团化学试剂有限公司(中国上海);甲基丙烯酸(MAA)、乙二醇二甲基丙烯酸酯(EGDMA,纯度>98%)、3-(甲基丙烯酰氧)丙基三甲氧基硅烷(3-methacryloxypropyltrimethoxysilane,MPS) 购自J&K Chemicals(中国上海),所有试剂均为分析级。
HH-A数显恒温水浴锅 常州市英格尔仪器制造有限公司;傅里叶变换红外光谱仪 美国Thermo Scientific公司;UV-1801紫外/可见分光光度计 北京瑞利分析仪器公司。
1.2 实验方法
1.2.1 制备羧基化葡聚糖修饰的Fe3O4磁性纳米粒子(CM-Fe3O4) 葡聚糖包覆磁性纳米粒子的制备采用共沉淀法[9]。将4 g葡聚糖和2 g六水合氯化铁溶解在30 mL的去离子水中,加热搅拌使葡聚糖完全溶解,加入0.8 g四水合氯化亚铁,15 min后,通入N2保护,迅速加入5 mL氨水,剧烈搅拌。升温至60 ℃搅拌1 h后,冷却至室温,以7000 r/min的速度离心20 min,除去大的团聚颗粒。透析袋(MWCO:14000)透析24 h后将得到的葡聚糖修饰的磁流体(dex-Fe3O4)放入250 mL三颈瓶中,加入4 g氢氧化钠,再缓慢地加入4 g氯乙酸,通N2保护,将混合溶液置于70 ℃水浴中,100 r/min机械搅拌90 min。反应完成后,冷却至室温,透析24 h,再放入0.1 moL·L-1的稀盐酸溶液中透析24 h,最后用去离子水透析24 h,磁流体真空干燥一夜,得到羧基化葡聚糖修饰的Fe3O4磁性纳米粒子(CM-Fe3O4)。
1.2.2 制备硅烷偶联剂修饰的Fe3O4磁性纳米粒子(Fe3O4@SiO2-MPS) 自制Fe3O4(0.5 g)分散在60 mL异丙醇:超纯水(5∶1,V/V),超声20 min。持续搅拌下,加入10 mL氨水和4 mL四乙氧基硅烷(Tetraethoxysilane,TEOS)。将混合溶液在室温下搅拌12 h。反应结束后,通过外部磁场收集磁性粒子(Fe3O4@SiO2),用超纯水多次洗涤,60 ℃真空干燥24 h。将干燥后的纳米粒子分散在含有5 mL MPS的50 mL无水甲苯中,超声分散15 min后,N2保护下70 ℃反应24 h。磁铁收集产物,经乙醇和去离子水洗涤后干燥备用[10]。
1.2.3 制备酪胺磁性分子印迹聚合物 将模板分子酪胺(0.1 mmol)和功能单体MAA(0.4 mmol)分散在4 mL乙腈中,并将混合物在4 ℃下反应12 h,CM-Fe3O4和Fe3O4@SiO2-MPS(100.0 mg)分别加入上述混合液中搅拌2 h,加入交联剂EGDMA(2.0 mmol)和引发剂AIBN(20.0 mg),并用N2吹扫5 min以除去氧气。然后60 ℃下反应24 h。聚合之后,以甲醇-乙酸(9∶1,v/v)为洗脱液索氏提取24 h,再用甲醇洗涤三次,干燥并研磨过筛得到dex-MMIPs和MPS-MMIPs,同时在不存在模板分子的情况下用相同的步骤制备dex-MNIPs(magnetic non-molecularly imprinted polymers)和MPS-MNIPs。
1.2.4 不同磁性载体的印迹聚合物动态吸附实验 为了评估制备聚合物的结合动力学,将20 mg聚合物分散在10.0 mL酪胺-甲醇溶液(5 mg·L-1)中,25 ℃水浴振荡0、5、10、15、20、25、30、60和90 min。随后,通过外部磁铁分离混合物,上清液过0.45 μm滤膜后通过UV在226 nm处测定。吸附容量Q(mg·g-1)用下式计算:
式(1)
式中,C0为酪胺溶液的初始浓度(mg·L-1),C1为吸附后上清液中酪胺浓度(mg·L-1),v为初始酪胺溶液体积(mL),m是聚合物的重量(g)。通过伪二阶方程进行分析[11],公式如下
式(2)
式中,Qt是t时刻的吸附量(mg·g-1),Qe是平衡时的吸附量(mg·g-1),k二阶吸附常数(mg·g-1·s-1)。
1.2.5 不同磁性载体的印迹聚合物静态吸附实验 为了评估聚合物对酪胺的结合能力,将20.0 mg聚合物加入到10.0 mL不同浓度(0、1、5、10、15、20、25和30 mg·L-1)的酪胺-甲醇溶液中,25 ℃振荡20 min。用同样方法计算吸附量Q(mg·g-1)。并且通过Scatchard模型对吸附结果进行分析,公式如下所示:
式(3)
式中,Qm是最大吸附容量(mg·g-1),Kd是Scatchard常数。
1.2.6 特异性吸附实验 将VPP作为TM的结构类似物参与到聚合物的选择性吸附实验中。分别取20 mg聚合物加入5 mg·L-1的结构类似物(VPP)溶液中,混合物在25 ℃下振荡20 min。外部磁铁分离,过滤并分析计算吸附量。其中两种化学物质的结构如图1所示。
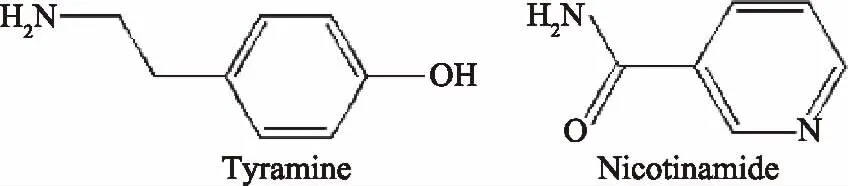
图1 结构类似物的化学结构Fig.1 Structure of analogs
2 结果与分析
印迹聚合物的制备过程如图2所示。首先,不同功能基团修饰的Fe3O4磁性纳米粒子的制备,其次是MIPs在磁性粒子表面的聚合,最后用合成的磁性印迹聚合物吸附模板分子。其中CM-Fe3O4通过两步法合成。第一步通过共沉淀法制备涂覆有葡聚糖的Fe3O4。第二步,Fe3O4表面上的葡聚糖与氯乙酸反应,在纳米粒子表面引入-COONa最后酸化得到-COOH。Fe3O4@SiO2-MPS的合成同样也是采用两步法,首先用TEOS使Fe3O4表面涂覆SiO2层,然后通过引入MPS使双键被接枝在Fe3O4@SiO2表面。将得到的两种不同材料修饰的磁性纳米粒子,TM,MAA和EGDMA聚合获得酪胺的磁性分子印迹聚合物。
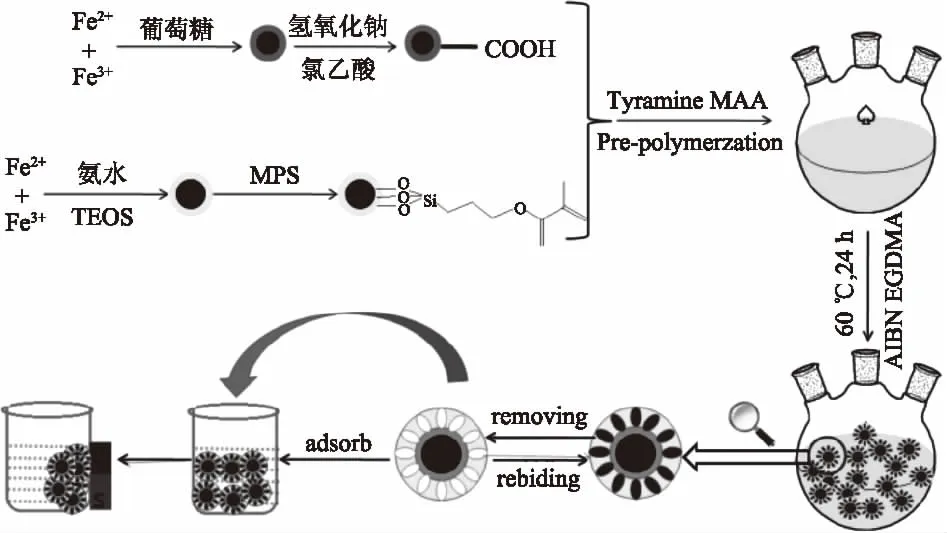
图2 酪胺磁性分子印迹聚合物合成示意图Fig.2 Schematic illustration of the process for preparation of tyramine magnetic molecularly imprinted polymers
2.1 FT-IR分析
图3为几种粒子的红外谱图。其中580 cm-1的峰来自Fe-O的振动。图3a中,1100和800 cm-1附近的峰为Si-O的不对称伸缩振动和对称伸缩振动,480 cm-1作为Si-O弯曲振动,表明SiO2成功地修饰Fe3O4磁性纳米粒子,同时2977 cm-1表示=CH2的对称伸缩振动,证明双键已经成功的修饰在Fe3O4@SiO2的表面[12]。图中3(b、c、d)的3400、2927和1013 cm-1处的峰是葡聚糖结构中存在的-OH,-CH2和C-O-C键的特征峰。此外,在样品c中1598 cm-1的峰是羧酸盐中-CO2的伸缩振动,样品d中1735 cm-1处的吸收带归因于游离羧酸中C=O伸缩振动峰。这说明葡聚糖分子已经成功连接到磁性纳米粒子表面,并完成羧酸化。

图3 红外光谱图Fig.3 FT-IR spectra注:(a):Fe3O4@SiO2-MPS,(b):dex-Fe3O4,
2.2 XRD分析
图4显示了(a)Fe3O4、(b)Fe3O4@SiO2-MPS和(c)CM-Fe3O4的XRD图。在20°~70°范围内有六个相对较强的衍射峰是220°、311°、400°、422°、511°和440°,与JCPDS数据库中的Fe3O4匹配良好(JCPDS卡:19~629)[13]。其中a,b,c三种物质的衍射峰并没有明显的差别,说明在修饰过程中Fe3O4核心晶体结构并没有发生变化。
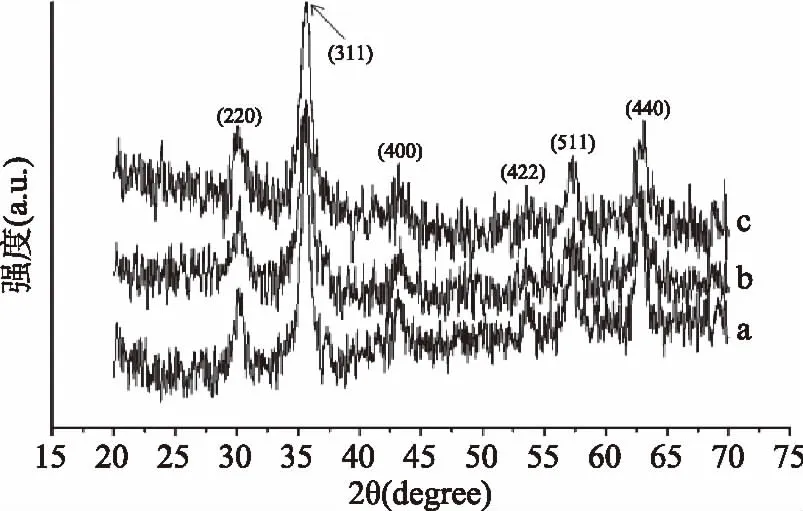
图4 聚合物的XRD图Fig.4 XRD of polymers注:(a):Fe3O4,(b):Fe3O4@SiO2-MPS,(c):CM-Fe3O4。
2.3 TEM分析
通过透射电镜对合成的磁性粒子进行观察,结果如图5所示。dex-Fe3O4(b)与未经修饰的Fe3O4(a)相比,分散性更好,粒径变大,这可能是由于未经修饰的Fe3O4因为巨大的表面能导致团聚严重,而长链葡聚糖分子增加了颗粒之间的空间位阻从而改善磁性纳米颗粒的分散性。

图5 磁性粒子的透射电镜Fig.5 TEM of magnetic nanoparticles注:(a):Fe3O4,(b):dex-Fe3O4。
2.4 不同磁性载体的印迹聚合物动态吸附实验
聚合物对酪胺的吸收动力学结果如图6所示。dex-MMIPs对酪胺的吸附量在前10 min内迅速增加,在20 min左右迅速达到吸附平衡,MPS-MMIPs在60 min左右才能达到吸附平衡,而很多分子印迹聚合物对目标物达到吸附平衡一般要很长时间,如杨眉等人制备的链霉素分子印迹聚合物达到吸附平衡所需要的时间约为16 h[14],周兴农等[2]制备的酪胺分子印迹聚合物吸附平衡需要8 h。同时我们制备的MPS-MMIPs的吸附量明显低于dex-MMIPs,可能是葡聚糖的表面含有大量的活性基团,增加了有效印迹位点的形成,此外传质效率的提高,也有助于模板分子的洗脱,使印迹聚合物形成更多的特异性结合位点,使得dex-MMIPs的吸附量高于MPS-MMIPs。以上结果说明制备的dex-MMIPs传质效率和吸附量有很大的提高,更能满足日益发展的社会对于物质前处理的要求。
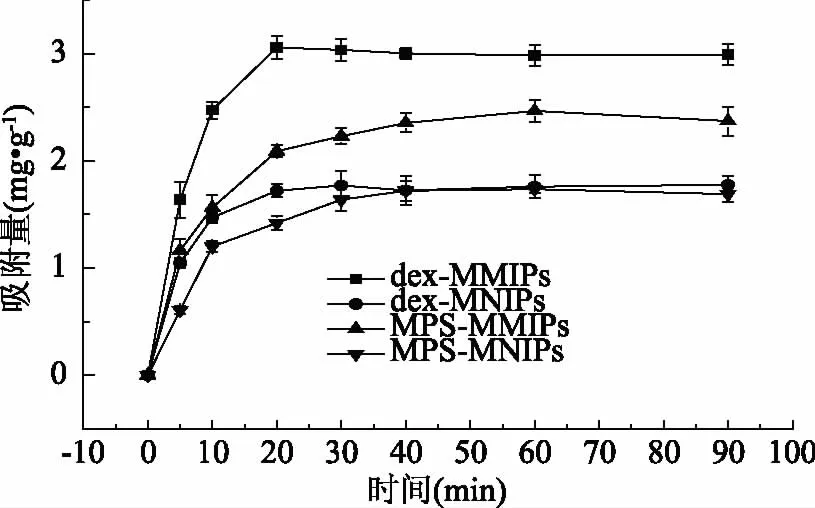
图6 对酪胺的动态吸附实验Fig.6 Dynamic binding experiments for TM
对四种聚合物的动力学曲线进行伪二级动力学分析[15],得出的结果如表1所示。其中R2均大于0.99说明伪二级动力学方程线性拟合较好,理论值得到的Qe分别为3.09、1.83、2.56和1.86 mg·g-1,与实验值3.05、1.72、2.08和1.41 mg·g-1(图6)符合,证明符合伪二级动力学方程。

表1 聚合物伪二级动力学模型参数Table 1 Parameters of pseudo-second-order model of polymers
2.5 不同磁性载体的印迹聚合物静态吸附实验
聚合物对酪胺的吸附量都随着浓度的增加而增加,当浓度达到20 mg·L-1时吸附速度减慢,这是由于聚合物对酪胺的吸附接近饱和。dex-MMIPs对TM的吸附量总是高于MPS-MMIPs(图7),这可能是因为葡聚糖表面的众多基团,使得在磁性粒子表面形成的印迹聚合物层很薄,同时传质速率的提高,使得模板分子洗脱的更彻底,表面的特异性孔穴增多,相比于MPS-MMIPs解决了包埋过深,洗脱不完全导致的吸附量不高的问题。两种非印迹聚合物的吸附量则相差不大,因为不存在模板分子的问题,都是非特异性的吸附位点,故而没有明显区别。
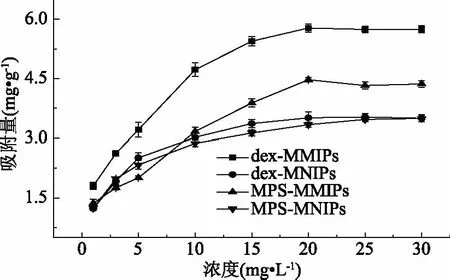
图7 对酪胺的静态吸附Fig.7 Static binding isotherms for TM
图8是对聚合物进行Scatchard分析,印迹聚合物通过Q/C对Q作图可以明显看出呈现非线性关系,存在两种吸附曲线,表明存在两种不同的结合位点[16]。通过拟合可以得出dex-MMIPs和MPS-MMIPs的髙亲和力位点方程分别为y=-0.159x+1.1794和y=-0.092x+0.5974,计算得到Qm分别为7.4和6.4 mg·g-1证明dex-MMIPs比MPS-MMIPs吸附性能方面呈现一定的优势。

图8 聚合物的Scatchard分析Fig.8 Scatchard analysis of polymers
2.6 特异性吸附实验
从图9可以看出,dex-MNIPs和MPS-MNIPs对两种物质的吸附量都不高,并且差异不显著,说明非印迹聚合物对物质的吸附只是非特异性吸附,在合成过程中没有模板分子的存在,并没有形成特异性的孔穴,但是dex-MNIPs相比MPS-MNIPs的分离效果要更好,是因为模板分子洗脱更完全,留下了更多的特异性位点,在本实验中对烟酰胺和酪胺的分离效果不是特别好,推测原因是两种物质的结构类似,同样存在可能产生氢键的O和N的影响。

图9 聚合物对酪胺(TM)和烟酰胺(VPP)的选择性Fig.9 Selectivity of polymers for Tyramine(TM)and Nicotinamide(VPP)
3 结论
以酪胺为模板分子,将表面分子印迹和磁性纳米技术相结合,以羧甲基葡聚糖包被的磁性纳米粒子为新型载体,成功制备酪胺的dex-MMIPs,通过与硅烷偶联剂修饰磁性纳米粒子为载体制备的MPS-MMIPs进行比较,dex-MMIPs在20 min左右达到吸附平衡,而MPS-MMIPs则至少需要60 min,表明葡聚糖修饰磁性微球具有优于硅烷偶联剂修饰磁性微球的传质性能。本文为酪胺预处理提供了一种新的传质效率高,稳定性好的表面分子印迹方法,为后期在食品中的应用提供了基础性研究。
