涎酸贮积症一家系报告
2017-07-31季涛云张月华包新华
季涛云 张 尧 张月华 包新华
北京大学第一医院儿科(北京 100034)
涎酸贮积症一家系报告
季涛云 张 尧 张月华 包新华
北京大学第一医院儿科(北京 100034)
目的探讨涎酸贮积症的临床特点及致病基因。方法回顾分析一家系涎酸贮积症的临床资料及基因检测结果。结果先证者为13岁女童,7岁时出现肢体疼痛,随后出现进行性视力下降及抽搐发作;有共济失调体征;眼底检查提示视神经萎缩;视诱发电位显示双眼P100潜伏期明显延长。先证者哥哥有类似表现。应用PCR方法对NEU1基因的外显子及其外显子-内含子连接区域进行扩增,采用DNA直接测序对此基因行突变检测。发现先证者及其哥哥均携带NEU1基因c.239C>T(p.P80L)和c.544A>G(p.P80L)复合杂合突变,分别来自其表型正常母亲和父亲,均为已报道的致病突变。结论明确涎酸贮积症家系的NEU1基因致病突变。
涎酸贮积症; 癫痫; 视力下降; 共济失调;NEU1基因突变
涎酸贮积症由Durand等于1977年首先命名,是一种罕见的常染色体隐性遗传的溶酶体病[1]。其致病基因为NEU1,此基因突变导致唾液酸苷酶(neuraminidase)活性降低,引起唾液酸化的复合糖分解途径缺陷,使其在溶酶体中贮积,从而产生一系列临床表型,如面容丑陋,肝脾肿大,进行性视力下降、肌阵挛、小脑性共济失调等。中国大陆地区鲜见相关病例报道。现将确诊的涎酸贮积症一家系报告如下。
1 临床资料
先证者,女,13岁,因“肢体疼痛6年余,视力下降4年余,间断抽搐15天”入院。患儿以肢体疼痛起病,表现为一侧或双侧小腿疼,多在劳累和遇冷时出现,严重时睡眠中可疼醒,1分钟左右缓解,3~4天出现1次。发病后不久出现单侧肢体(左右不固定)节律性抖动,入睡后不久出现,每晚发作3~4次,每次持续数秒钟。4年前患儿出现视物模糊,之后呈进行性下降。3年前因肢体节律性抖动于外院诊断癫痫予左乙拉西坦治疗无明显效果。15天前出现抽搐发作,表现为全面性强直-阵挛发作,持续2分钟左右自行缓解,共出现2次。患儿既往体键,围生期无特殊,体型消瘦。患儿哥哥现29岁,10岁出现大腿疼痛和阵发性肢体抖动,后逐渐出现行走时步态拖曳,易摔跤。12岁出现视力下降,呈进行性下降;15岁左右瘫痪,现仍有阵发性肢体抖动,仅能在10 cm左右看到手动,听力及智力正常。当地诊断“癫痫,肌阵挛发作?”,未予特殊治疗。父母、姐姐(31岁)、双胞胎妹妹(非同卵)身体健康。入院体格检查:体型消瘦,身高141 cm,体质量25 kg,头围50 cm,视物模糊,双眼视力均为0.1,心、肺、腹查体未见异常,四肢肌力及肌张力正常,指鼻不准,走直线欠稳,四肢腱反射对称引出,病理征及脑膜刺激征阴性。实验室检查:血、尿、粪常规,肝、肾功能以及电解质、血糖、碳酸氢根、肌酶均无异常;乳酸、β-羟丁酸、同型半胱氨酸、血氨及血尿串联质谱分析未见异常;甲状腺功能、甲状腺抗体、皮质醇、生长激素、胰岛素生长因子无异常。视诱发电位:双眼分别图形翻转刺激,双侧波形不规则呈多切迹、低波幅状,基线漂移,其P100潜伏期均明显延长,左191.3 ms,右185.0 ms,波形分化及重复性较差。眼底检查:视神经萎缩,余未见明显异常。头颅磁共振成像正常。脑电图病初正常,3年后复查双侧Rolandic区为主棘波,快波活动,多棘波,多棘慢波发放,监测到左下肢肌电短暂爆发不伴有EEG改变。
经北京大学第一医院临床研究伦理委员会审批,患儿家属签署知情同意书后,采集包括先证者、其哥哥及父母等家系成员的外周血标本,盐析法提取外周血白细胞基因组DNA。以PCR引物作为测序引物,用末端终止法在ABI 9700型热循环仪上进行测序反应,反应结束后,延伸产物在ABI PRIS M3730型XL DNA序列分析仪上进行分析,对测序异常的片段重新进行PCR扩增,再次正、反向测序,以验证结果的可靠性。测序结果应用DNA Star软件包中的SeqMan(Lasergene公司产品)软件进行序列对比分析。据序列分析结果查阅基因突变数据库,若为尚未报道过的新突变,通过分析氨基酸变化、保守性,已知基因多态性数据库检索及与50例健康成人序列对照,以确定所发现的突变为致病突变,排除基因多态性。若为已知致病突变或新突变已排除了基因多态性,则进一步对其父母进行基因扩增测序,确定突变来源。
NEU1基因检测结果:患儿及其哥哥有相同的复合杂合突变,分别为c.239C>T(p.P80L)和c.544A>G(p.S182G),见图1。c.239C>T和c.544A>G分别来自表型正常的母亲和父亲。患儿姐姐和妹妹未进行基因检测。
2 讨论
涎酸贮积症起病年龄不同,表型亦不同。1979年,Lowden 等[2]根据表型的不同将其分为Ⅰ型和Ⅱ型。涎酸贮积症Ⅰ型起病晚(多见于10~20岁),症状轻,无体表及内脏畸形,其典型的表现包括进行性视力损害、肌阵挛、小脑性共济失调,无或仅有轻微智力损害,眼底检查可发现樱桃红斑,因此,Ⅰ型也称为樱桃红斑肌阵挛综合征。涎酸贮积症Ⅱ型发病较Ⅰ型早、临床表现亦更为严重。根据起病年龄不同分为3型,分别为先天型(宫内起病)、婴儿型(1岁内发病)和少年型(2岁后发病)。涎酸贮积症Ⅱ型常伴面容丑陋,肝脾肿大、骨骼异常(如骨骼发育不全、椎骨畸形)、严重的智力障碍等,先天型常表现为胎儿水肿。
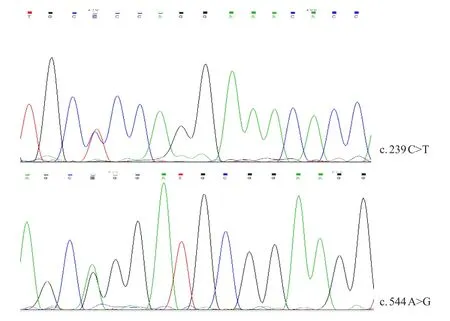
图1 先证者NEUT1基因测序图
本例患儿临床特点为:①女性13岁,慢性病程,逐渐进展;②以肢体疼痛起病,后出现进行性视力下降,肢体抖动,癫痫发作及行走不稳的表现;③自幼体格发育落后,运动耐力差,智力发育可;④家族史:患儿哥哥有类似表现;⑤体格检查:体型消瘦,共济失调(指鼻试验及走直线欠稳);⑥辅助检查:眼科视神经经萎缩,视诱发电位P100潜伏期均明显延长,脑电图Rolandic区为主癫痫放电。根据患儿的症状、体征及辅助检查有多系统受累包皮层,视神经及小脑。患儿起病后表现为慢性病程,缓慢进展,神经系统多部位受累,结合家族史,遗传性疾病尤其是神经变性病可能性大。结合典型的进行性视力损害、癫痫发作、小脑性共济失调和基因检测结果,最终明确诊断为涎酸贮积症Ⅰ型。
患儿及其哥哥携带NEU1基因c.239C>T(p.P80L)和c.544A>G(p.P80L)复合杂合突变,分别来自其表型正常母亲和父亲,此两个突变均为已报道的致病突变。c.544A>G可能是中国人较为常见的热点突变。在中国台湾地区,17例确诊唾液酸沉积症Ⅰ型病例均携带c.544A>G(S182G)突变,其中15例为纯合突变,另2例为复合杂合突变[3]。另2例中国大陆报道的唾液酸沉积症Ⅰ型患者也均有此位点的突变[4,5]。研究发现,在COS-7细胞中表达含有S182G突变的NEU1基因,唾液酸苷酶的活性为正常的20%~40%,有缺陷的酶可被运送至溶酶体,形成多酶复合物并稳定存在[5]。另一个突变位点c.239C>T所编码的氨基酸(P80)位于唾液酸苷酶蛋白第1个β折叠单元的保守FRIP模体内,此氨基酸能引起较大的主链构象改变,从而影响唾液酸苷酶的活性[6]。
眼底樱桃红斑是涎酸贮积症Ⅰ型的特征性表现,也可在Ⅱ型中出现[7]。有文献报道在涎酸贮积症Ⅰ型患者中的发生率为100%[2]。亦有报道部分患者可延迟在发病后15年发现眼底樱桃红斑[8]。曾有报道1例15岁时以肌阵挛起病的唾液酸沉积症Ⅰ型患者,35岁首次观察到眼底樱桃红斑[9]。本例患儿眼底检查未发现樱桃红斑,不除外与病程尚短有关,仍需继续随访。进行性视力下降为唾液酸沉积症Ⅰ型另一常见表现,主要由视网膜及视神经病变引起。部分患者视力下降不突出,而早期出现视诱发电位的异常。在中国台湾地区的17例唾液酸沉积症Ⅰ型患者中,16例(94.1%)视觉诱发电位异常,仅4例(23.5%)有视力下降的临床症状,因此视觉诱发电位检查具有早期诊断价值[3]。
除以上两点之外,涎酸贮积症Ⅰ型常伴有肌阵挛、共济失调、癫痫发作等症状[10],需要与几种可致进行性肌阵挛癫痫的常见病因进行鉴别,如神经元蜡样脂褐质沉积症、线粒体脑肌病如MERRF、Lafora病、戈谢病等[11,12],但与以上疾病相比涎酸贮积症Ⅰ型智力影响较轻。唾液酸沉积症尚无可靠治疗方法[13],临床治疗也多限于对症治疗。如癫痫的治疗,文献报道氯硝西泮、丙戊酸等用于治疗此病的肌阵挛有较好的效果[3]。
本研究通过对一个家系的NEU1基因研究分析,明确了该家系中的致病基因突变及相关个体基因状况,为该家庭进行准确的遗传咨询提供了可能。
[1]Durand P, Gatti R, Cavalieri S, et al. Sialidosis (mucolipidosis I) [J]. Helv Paediatr Acta, 1977, 32(4-5): 391-400.
[2]Lowden JA, O'Brien JS. Sialidosis: a review of human neuraminidase defciency [J]. Am J Hum Genet, 1979, 31(1):1-18.
[3]Lai SC, Chen RS, Wu Chou YH, et al. A longitudina l study of Taiwanese sialidosis type 1: an insight into the concept of cherry-red spot myoclonus syndrome [J]. Eur J Neurol, 2009, 16(8): 912-919.
[4]张包静子, 全 超, 罗苏珊, 等. 眼底樱桃红斑(唾液酸沉积症Ⅰ型1 例报道及文献复习)[J]. 中国临床神经科学,2015, 23(1): 115-120.
[5]Lukong KE, Landry K, Elsliger MA, et al. Mutations in sialidosis impair sialidase binding to the lysosomal multienzyme complex [J]. J Biol Chem, 2001, 276(20):17286-17290.
[6]Itoh K, Naganawa Y, Matsuzawa F, et al. Novel missense mutations in the human lysosomalsialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes [J]. J Hum Genet, 2002, 47: 29-37.
[7]Canafoglia L, Franceschetti S, Uziel G, et al. Characterization of severe action myoclonus in sialidoses [J]. Epilepsy Res,2011, 94(1-2): 86-93.
[8]Palmeri S, Villanova M, Malandrini A, et al. Type I sialidosis:a clinical, biochemical and neuroradiological study [J]. Eur Neurol, 2000, 43(2): 88-94.
[9]Michalewska Z, Gajos A, Michalewski J, et al. Spectral optical coherence tomography in a patient with type I sialidosis [J]. Med Sci Monit, 2011, 17(10): CS129-131.
[10]Canafoglia L, Robbiano A, Pareyson D, et al. Expanding sialidosis spectrum by genome-wide screening:NEU1mutations in adult-onset myoclonus [J]. Neurology, 2014,82(22): 2003-2006.
[11]Lamperti C, Zeviani M. Myoclonus epilepsy in mitochondrial disorders [J]. Epileptic Disord, 2016,18(S2): 94-102.
[12]Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses [J]. Epileptic Disord, 2016, 18(S2): 73-88.
[13]d'Azzo A, Machado E, Annunziata I. Pathogenesis, Emerging therapeutic targets and Treatment in Sialidosis [J]. Expert Opin Orphan Drugs, 2015, 3(5): 491-504.
Sialidosis: a case report
JI Taoyun, ZHANG Yao, ZHANG Yuehua, BAO Xinhua (Department of Pediatrics, Peking University First Hospital, Beijing 100034, China)
ObjectiveTo explore the clinical features and pathogenic genes of sialidosis.MethodsThe clinical data and genetic test results of a family with sialidosis were retrospectively analysed.ResultsThe proband was a 13-year-old girl who presented with limb pain at age 7, followed by progressive vision loss and convulsive seizure. In addition, she also had the sign of ataxia. Fundus examination showed optic atrophy in her eyes. Visual evoked potential showed that the latency of binocular P100 was signifcantly prolonged. The elder brother of the proband showed similar manifestation. PCR was used to amplify the exons and exon-intron boundaries of theNEU1gene, and DNA direct sequencing was used to detect the mutation in this gene. It was found that both proband and her brother carried two known pathogenic heterozygous mutations in the NEU1 gene, c.239C>T(p.P80L) and c.544A>G (p.P80L) respectively from both their mother and father of normal phenotype.ConclusionThe causative mutation of theNEU1gene in the family of sialidosis has been defned.
sialidosis; epilepsy; decreased vision; ataxia;NEU1gene mutation
2016-10-08)
(本文编辑:梁 华)
10.3969/j.issn.1000-3606.2017.07.014
包新华 电子信箱:zwhang@pku.edu.cn
