Shwachman-Diamond综合征患儿的肝脏病理和基因分析
2017-07-31欧阳文献谭艳芳李双杰
姜 涛 欧阳文献 谭艳芳 李双杰
湖南省儿童医院肝病中心(湖南长沙 410007)
Shwachman-Diamond综合征患儿的肝脏病理和基因分析
姜 涛 欧阳文献 谭艳芳 李双杰
湖南省儿童医院肝病中心(湖南长沙 410007)
目的分析Shwachman-Diamond综合征(SDS)的基因异常及肝脏病理。方法回顾分析1例SDS患儿的临床资料。结果患儿,男,1月龄起病,以中性粒细胞减少为首发表现,伴贫血、转氨酶升高、反复感染,而胰腺外分泌功能障碍症状不典型。肝脏穿刺术病理学检测,光镜示肝细胞轻度损害。采集患儿及父母血标本,采用二代基因测序检测发现SBDS(NM_016038.2) Intron2 c.258+2T>C p.?纯合突变,突变来源于父母亲。结论基因检测有助于确诊SDS,有条件者可行肝脏穿刺术。
Shwachman-Diamond综合征; 肝脏病理; 基因
儿童胰腺功能不全并中性粒细胞减少综合征也称Shwachman-Diamond综合征(Shwachman-Diamond sydrome,SDS),是一种少见的常染色体隐性遗传病(OMIM 260400),患病率为1/20 000~1/10 000,系SBDS等位基因突变导致,特点是胰腺外分泌功能不全,骨骼异常,血液系统异常,且有发展成骨髓增生异常综合征(myelodysplastic syndrome,MDS)和再生障碍性贫血的风险[1]。至今我国确诊的病例少见。本研究通过基因检测确诊1例SDS患儿,现将其临床表现,肝脏病理学检测和基因检测结果报告如下,以提高临床医师对该病的了解。
1 临床资料
患儿,男,1岁3个月。因发现中性粒细胞减少及转氨酶升高1年余住院。患儿出生后1个月即出现中性粒细胞减少,2月龄时出现肝功能受损和贫血,贫血严重时血红蛋白为53 g/L,予以输血、护肝、降酶等治疗后贫血纠正,复查肝功能和中性粒细胞仍异常,一直无腹泻等不适,在外院行骨穿确诊单纯红细胞再生障碍性贫血。既往体质欠佳,平时易患呼吸道感染,有高胆红素血症,听力受损病史,2月龄时曾感染结核,予以异烟肼及利福平抗结核治疗3个月。个人史及家族史无特殊。入院体格检查:发育正常,营养略欠佳,急性面容,心肺无异常,肝脏未触及,脾脏未触及,四肢肌力正常、肌张力正常,病理征阴性。实验室检查:病程中患儿血常规中性粒细胞波动在(0.28~1.42) ×109/L;谷氨酸氨基转移酶波动在52~695 U/L,天冬氨酸氨基转移酶波动在100~487 U/L;输血全套和肝炎全套正常,EB病毒抗体,巨细胞病毒DNA荧光定量阴性,甲状腺功能检查正常,血串联质谱筛查和尿气相色谱质谱正常,肝纤维四项(血清Ⅲ型胶原、血清层粘连蛋白、血清透明质酸酶、Ⅳ型胶原蛋白)测定提示肝纤维化,地中海贫血基因阴性,甲胎蛋白和铜蓝蛋白正常;血淀粉酶、脂肪酶正常。腹部B超:肝门区淋巴结稍大,胰腺回声增强。左手X线片示骨龄落后1岁;四肢X片示四肢长骨骨质疏松,骨皮质变薄。
经家长知情同意和医院伦理委员会核准,对患儿进行肝脏穿刺术和基因检测。在静脉麻醉下行肝脏穿刺术,病理示光镜下肝细胞轻度损害(轻度炎症和纤维化,即G1S1),见图1。用Sanger双脱氧链终止法检测示SBDS(NM_016038.2) Intron2 c.258+2T>C p.?纯合突变,致病突变,其父母亲均有SBDS(NM_01603 8.2) Intron2 c.258+2T>C p.?杂合突变,见图2。
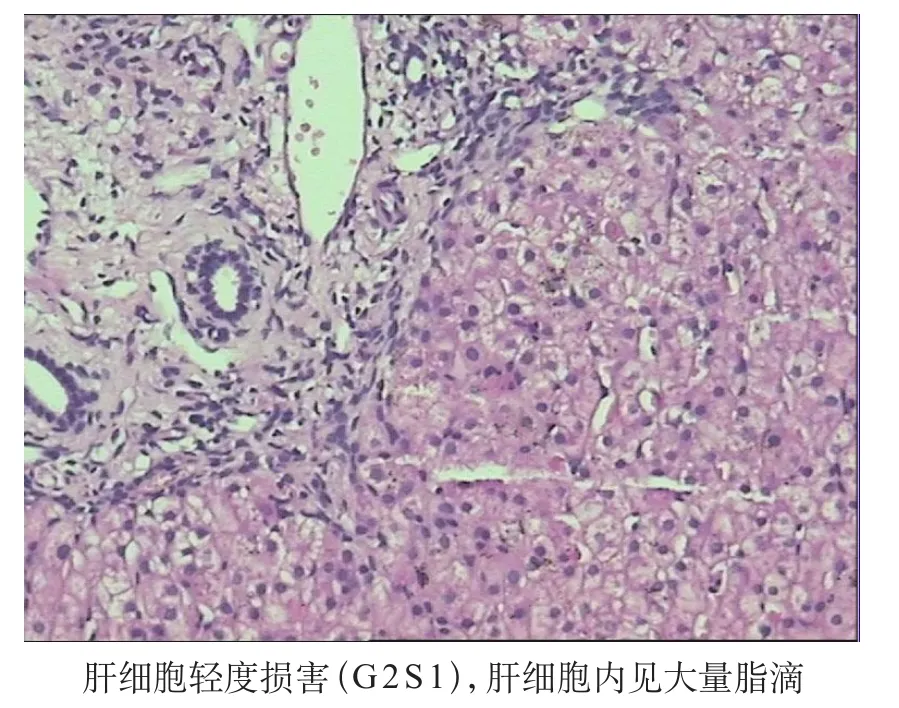
图1 肝脏光镜检查(×400)
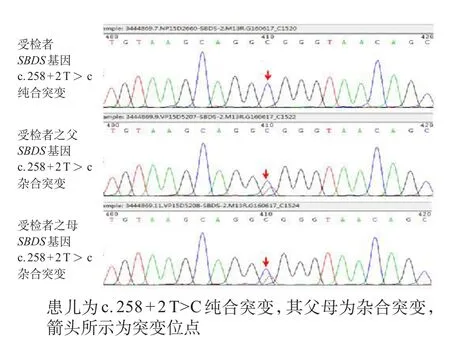
图2SBDS基因检测
确诊后患儿给予对症支持治疗,一直予以双环醇降酶,水飞蓟宾葡甲胺和复方甘草酸酐护肝,补充脂溶性维生素等治疗,随访1年余,患儿仍有反复呼吸道感染,多次复查肝功能和中性粒细胞仍异常。
2 讨论
SDS的经典临床表现为胰腺外分泌功能障碍与血细胞系统异常,大多数功能障碍一般在出生1年内出现,往往是前6个月,胰腺腺泡细胞严重枯竭导致胰腺外分泌功能障碍,主要表现为营养吸收不良,脂肪泻和生长发育落后。随着年龄的增长,50%的患者4岁时在不补充胰酶的情况下能正常吸收脂肪[2]。血液学表现包括间歇性或持续性血细胞减少[3],有的甚至是首发表现,中性粒细胞减少和中性粒细胞趋化作用损伤可能是幼儿反复感染的最重要的因素[4],急性和深部组织感染可能危及生命[5]。SDS患者发生全血细胞减少的重型再生障碍性贫血,骨髓增生异常综合征(MDS)的风险或进展为急性髓系白血病(AML)的风险明显增加,MDS/AML的发生被认为是终生的,但对其具体发病年龄现在的意见不统一[6,7]。另外,肝脏肿大,肝功能异常,骨骼特征变化(如软骨发育异常或先天性胸萎缩症)也比较常见,有的甚至以神经肌肉疾病[8]、肝脾肿大为首发表现[9]。
SBDS基因(OMIM 607744)定位于染色体7 q 11.21,针对外显子的3种常见的SBD致病突变位点,c.183_184delinsCT,c.258+2T>C,和c.[183_184delinsCT:258+2T>C ]的分析显示,约90%患者至少有一个致病突变,62%有多个致病突变[10],约10%的临床诊断患者中无此突变,其诊断依赖于临床表现,包括胰腺功能外分泌障碍证据和骨髓衰竭与一系或多系血细胞减少[11,12]。随诊基因检测技术的发展,更多的表现谱被发现,Myers等[13]研究显示37例基因确诊的SDS患者中,有一半没有经典的临床表现。本研究中,患儿以中性粒细胞减少为首发表现,伴贫血,转氨酶升高,易反复感染,而胰腺外分泌功能障碍症状不典型,基因检测示c.258+2T>C纯合突变,为常见的基因突变,基因来源于父母亲。由此可见,如果按经典的临床表现来诊断容易导致漏诊,因此对临床上怀疑SDS的患者应尽早基因检测。
SDS患者早期易合并有肝脏肿大和转氨酶升高,但SDS相关性肝病总体预后良好,一般在5岁以后趋于正常[14],而相关影像学和生化学亦无肝脂肪变性、肝硬化、或纤维化的证据,部分转氨酶水平正常化后有轻度胆汁淤积[15],有限的肝活检提示轻微至明显的肝脏病理学改变,其损伤为非进展性的。本研究发现患儿有转氨酶升高,持续2年余,肝脏病理学检查中光镜示肝脏组织结构稍紊乱,可见中央静脉,未见完整肝小叶结构。肝索结构尚清晰。胆小管发育尚可。肝细胞部分有水肿。胞核大小基本一致,以单个核细胞为主。个别肝细胞点状坏死。汇管区多灶炎症细胞浸润,肝细胞间少量炎症细胞浸润,炎症细胞以淋巴细胞为主,可见少数枯否细胞增生(G1)。纤维组织轻度增生,未见纤维间隔或假小叶结构。血管内皮细胞未见明显增生(S1)。
SDS目前的治疗主要是口服胰酶和脂溶性维生素[16]来补充胰外分泌胰腺功能不全。贫血,中性粒细胞和血小板减少症可考虑输入血小板和输血和粒细胞集落刺激因子,严重的全血细胞减少症,MDS或者AML者可考虑予以造血干细胞移植,同时应注意检测血常规,生长发育,营养状态,骨髓及骨关节,神经心理筛查等。
SDS的发病率低,某些患者的症状不典型及临床医师的认识不足,容易漏诊,故对于病因不明的反复肝功能和中性粒细胞异常的患儿,建议尽早行基因检测,有条件者可行肝脏穿刺术,以免延误治疗,并可严密检测其发展成MDS或AML的风险,同时为优生优育做出指导。
[1]Cho WK, Jung IA, Kim J, et a1. Two cases of Shwachman-Diamond syndrome in adolescents confirmed by genetic analysis [J]. Ann Lab Med, 2015, 35(2):269-271.
[2]Mack DR, Forstner GG, Wilschanski M, et a1. Shwachman syndrome: Exocrine pancreatic dysfunction and variable phenotypic expression [J]. Gastroenterology, 1996, 111(6):1593-1602.
[3]Donadieu J, Fenneteau O, Beaupain B, et a1. Classifcation of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome [J]. Haematologica, 2012, 97(9):1312-1319.
[4]Kuijpers TW, Alders M, Tool AT, et a1. Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype-phenotype relationship [J]. Blood, 2005,106(1):356-361.
[5]Grinspan ZM, Pikora CA. Infections in patients with Shwachman-Diamond syndrome [J]. Pediatr Infect Dis J,2005, 24(2):179-181.
[6]Hashmi SK, Allen C, Klaassen R, et a1. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation [J]. Clin Genet, 2011, 79(5):448-458.
[7]Tamary H, Nishri D, Yacobovich J, et a1. Frequency and natural history of inherited bone marrow failure syndromes:the Israeli Inherited Bone Marrow Failure Registry[J]. Haematologica, 2010, 95(8):1300-1307.
[8]Topa A, Tulinius M, Oldfors A, et a1. Novel myopathy in a newborn with Shwachman-Diamond syndrome and review of neonatal presentation [J]. Am J Med Genet A, 2016,170A(5):1155-1164.
[9]Wilschanski M, van der Hoeven E, Phillips J, et a1. Shwachman-Diamond syndrome presenting as hepatosplenomegaly [J]. J Pediatr Gastroenterol Nutr, 1994, 19(1):111-113.
[10]Boocock GR, Morrison JA, Popovic M, et a1. Mutations in SBDS are associated with Shwachman-Diamond syndrome[J]. Nat Genet, 2003, 33(1):97-101.
[11]Dror Y, Donadieu J, Koglmeier J, et a1. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome [J]. Ann N Y Acad Sci, 2011, 1242: 40-55.
[12]Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update [J]. Hematol Oncol Clin North Am, 2013, 27(1):117-128.
[13]Myers KC, Bolyard AA, Otto B, et a1.Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry [J]. J Pediatr, 2014, 164(4):866-870.
[14]Toiviainen-Salo S, Mäyränpää MK, Durie PR, et a1.Shwachman-Diamond syndrome is associated with lowturnover osteoporosis [J]. Bone, 2007, 41(6):965-972.
[15]Toiviainen-Salo S, Durie PR, Numminen K, et a1. The natural history of Shwachman-Diamond syndromeassociated liver disease from childhood to adulthood [J]. J Pediatr, 2009, 155(6):807-811.
[16]Pichler J, Meyer R, Köglmeier J, et a1. Nutritional status in children with Shwachman-diamond syndrome[J]. Pancreas, 2015, 44(4):590-595.
Liver pathology and gene analysis in children with Shwachman-Diamond syndrome
JIANG Tao, OUYANG Wenxian,TAN Yanfang, LI Shuangjie (Department of Hepatopathy Center, Hunan Children's Hospital, Changsha 410000, Hunan, China)
ObjectiveTo analyze the gene abnormality and liver pathology in Shwachman-Diamond syndrome (SDS) in a child.MethodThe clinical data of one child with SDS were analyzed retrospectively.ResultA male patient was 1 month old at onset with neutrophil decrease as the frst manifestation, accompanied by anemia, elevated transaminase and repeated infection.Exocrine pancreatic dysfunction was atypical. Pathological examination of liver biopsy showed slight damage of liver cells under light microscope. The blood samples of child and parents were collected, and homozygous mutations of SBDS (NM_016038.2)Intron2 c.258+2T>C p.? were detected by two generation gene sequencing. And these mutations were from both his parents.ConclusionGene testing is helpful in diagnosing SDS and we suggest that liver biopsy should be performed if condition allows.
Shwachman-Diamond syndrome; liver pathology; gene
2016-12-06)
(本文编辑:邹 强)
10.3969/j.issn.1000-3606.2017.07.017
李双杰 电子信箱:lesjie62@vip.sina.com
