睑裂狭小-上睑下垂-智障综合征1例报告并文献复习
2017-07-31曹丽芳童笑梅田亚萍宋
曹丽芳童笑梅田亚萍宋 琳
1. 北京大学国际医院儿科(北京 102206);2. 北京大学第三医院儿科(北京 100191)
睑裂狭小-上睑下垂-智障综合征1例报告并文献复习
曹丽芳1童笑梅2田亚萍1宋 琳1
1. 北京大学国际医院儿科(北京 102206);2. 北京大学第三医院儿科(北京 100191)
目的探索睑裂狭小-上睑下垂-智障综合征(BPID)的临床特征及基因突变。方法回顾新生儿重症监护病房(NICU)收治的1例BPID患儿的临床资料和诊治经过,结合PubMed数据库的检索文献,复习BPID及所属睑裂狭小-智障综合征(BMR)的常见分型、临床特点、诊断及遗传咨询。结果患儿出生胎龄39周,男,出生体质量1 920 g,生后15 min因呼吸困难收住NICU。主要临床表现为睑裂狭小、上睑下垂、小下颌等面部畸形,吸气性呼吸困难伴喉软骨软化、胸廓发育畸形,以及喂养困难等。通过全外显子基因测序,确定为UBE3B基因的复合杂合突变导致,诊断为BPID,为罕见基因病。查阅文献,国内尚未见相关报道,国外文献1篇含来自4个家庭的5例患者,属于BMR的分型之一,为常染色体隐性遗传病,均由UBE3B基因突变导致。结论BPID属BMR,临床罕见,全外显子基因测序可以明确诊断。
睑裂狭小; 上睑下垂; 智障; 基因突变
睑裂狭小-智障综合征(BMR),是以特殊面容和智障为最重要特征的少见疾病,1986年首次被报道和命名[1],BMR分型众多,是累及多系统的常染色体隐性遗传性疾病。而睑裂狭小-上睑下垂-智障综合征(BPID),是BMR的一个罕见分型,在2012年由Basel-Vanagaite 等[2]首次报道,目前,国外报道有4例,国内尚未见报道,已报道患者均因UBE3B基因突变引起,基因检测为确诊 BPID提供了直接证据。本病可引起生长发育、语言、运动和智力以及视听等多方面的落后和损害,患者生存能力差。现将北京大学国际医院诊断的1例 BPID的病例报告如下,并结合相关文献进行复习。
1 临床资料
新生患儿,男,生后15 min,因哭声细小,吸气性喉鸣和三凹征,睑裂狭小、上睑下垂、小下颌等面部畸形,以“足月小样儿、呼吸困难原因待查?”收住新生儿重症监护病房(NICU)。患儿出生胎龄39+2周,胎盘小,脐带细,出生体质量1 920 g,Apgar评分1分钟9分(呼吸-1分),5分钟及10分钟均10分。父亲38岁,母亲35岁,否认家族遗传性疾病,因患儿父亲精子畸形及活动度低下通过辅助生育技术受孕,孕前父母常规染色体检查均未见异常。母孕期合并妊娠期糖尿病,饮食控制血糖在正常范围,孕期行无创性DNA产前筛查,提示13-三体综合征、18-三体综合征和21-三体综合征均为低风险,非近亲婚配,家族成员中无类似疾病史。
入院查体:体温36.9℃,脉搏136次/min,呼吸43次/min,收缩压(SBP)/舒张压(DBP):74/43mmHg,平均动脉压54 mmHg,经皮血氧饱和度97%,头围30.5 cm(<P3),身长45 cm(<P3),体质量1.92 kg(<P3)。前囟2.5 cm×2.5 cm,头发稀疏,毛发浅淡,睑裂狭小,上睑下垂,睑裂上斜,眼窝深,眼距宽,耳位低,宽/低鼻梁,鼻孔前倾,窄面,小嘴,小下颌,腭弓高,无唇腭裂,后囟1 cm×1 cm,骨缝重叠。皮肤薄,干燥脱屑,无气促,吸气性喉鸣和轻度三凹征,胸廓前后径饱满,心腹查体未见异常,右侧睾丸未降至阴囊,手指足趾细长,手拇指外展困难,双足外翻,四肢肌张力低下,吸吮反射存在,吞咽反射未引出。
实验室检查示血常规、肝肾功能、电解质均正常。染色体核型分析示46,XY,核型正常。胸片示胸廓形状呈钟形。超声心动图示先天性心脏病-主动脉瓣二叶畸形。颅脑超声示脑发育落后,成熟度欠佳,胼胝体发育不良。纤维支气管镜示喉软骨软化。肺部CT+三维重建示声门下至胸廓入口处气管细,隆突以上气管局限性增宽,右上叶支气管略迂曲纤细;声门下颈前区域软组织显厚;胸廓前后径略增宽,胸椎略显后凸。
临床结合查体发现存在睑裂狭小、上睑下垂、小下颌、低耳位、足外翻、手指细长、胸廓畸形、气道狭窄、胼胝体发育不良和先天性心脏畸形等特点,出生即出现哭声细小、持续吐沫,吸气性喉鸣及三凹征并逐渐加重,吸吮反射存在,无吞咽动作,因喂养困难需要持续鼻饲喂养,临床首先考虑Pierre Robin序列征(Pierre Robin sequence,PRS)[3],但该综合征的典型表现为小下颌、舌后坠及腭裂等口面部畸形三联征,同时可伴有呼吸困难、喂养困难等临床症状,本患儿无腭裂,且有睑裂狭小、上睑下垂等特殊面容和其他PRS不具备的特征,不能单纯用PRS解释,而且PRS诊断亦并非最终诊断,因有接近50%的PRS仅作为其他多发畸形综合征的一部分而出现,PRS诊断可仅为发现其他多发畸形综合征的一个起点。同时根据患儿有喂养困难、生长迟缓、窄面、小嘴、手指细长,隐睾、肌张力低下等临床特点,也疑诊Prader Willi综合征[4],但Prader Willi综合征患儿在胎儿期至3岁的主要临床特点还包括颅形长,嘴角向下等,本患儿均无以上临床表现,且Prader Willi综合征也无本例患儿的睑裂狭小、上睑下垂等特殊面部特征和胸廓、心脏畸形,气道狭窄,胼胝体发育不良等并发问题。基于以上分析,为进一步明确诊断,经家长知情同意后,于生后3天对患儿及其父母进行单基因病全外显子组测序分析,共住院42天出院。门诊随访过程中,于生后56天检测回报发现UBE3B基因复合杂合突变c.2222(exon20)delG和c.2737C>T(exon25)。突变位点c.2222(exon20)delG来源于母亲,为杂合移码突变(图1),该突变会导致所有蛋白序列异常变化;突变位点c.2737C>T(exon25),来源于父亲,为无义突变(图2),该突变会导致蛋白翻译提前终止。测序结果提示为睑裂狭小-上睑下垂-智障综合征(BPID),为常染色体隐性遗传病。其中c.2222 (exon20)delG突变位点在人类孟德尔遗传数据库(OMIM)中未见报道,在单核苷酸多态性数据库(dbSNP)中无rs编号,可能为新发现的突变位点。本例患儿的照片和X线胸片见图3、4。患儿出院时仍需鼻饲胃管喂养,体质量2 670 g,头围32 cm,身长52 cm,三项指标均低于同龄儿P3。出院后其父母一直带患儿在我院儿科门诊随访,每周更换胃管并接受护理指导,生后6个月采用首都儿科研究所制订的《0~6岁小儿神经心理发育检查表》进行大运动、精细运动、适应能力、语言、社会行为等五个能区的检测。测评结果为发育商(DQ)68,智能评价为低下。
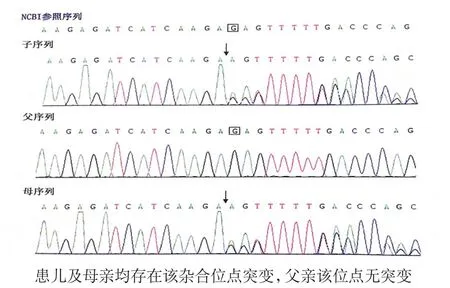
图1UBE3B基因第c.2222 (exon20)delG杂合位点突变
2 讨论
新生儿面部异常最易被发现,其中睑裂狭小较常见,睑裂狭小是指上、下眼睑垂直距离缩小、眼睑短小和紧绷,Guercio等[5]和Hall等[6]描述其他眶周畸形还包括:内眦赘皮、常伴发上睑下垂。Zlotogora等[7]认为这些眼部畸形是常染色体显性遗传病中睑裂狭小-上睑下垂-内眦赘皮综合征(blepharophimosisepicanthus inversus and ptosis syndrome,BPES)的常见症状。但临床除BPES外,睑裂狭小还可见于许多临床先天性畸形,尤其是Dubowitz综合征[8], Marden-Walker综合征[9]。1986年Ohdo等[1]首次在一对姐妹和她们的表弟身上将睑裂狭小和智障组合在一起,描述了一个新的常染色体隐性遗传的多重先天性畸形(Ohdo syndrome),也称为睑裂狭小-智障综合征(blepharophimosis-mental retardation syndromes,BMR)。之后又陆续报道了多个Ohdo类似综合征[10-14],扩大了BMR的病因和疾病谱。2006年Verloes 等[15]报告来自8个家庭的11例BMR,并提出一个分类方法,将BMR分为5种亚型:①del(3P)综合征,存在3号染色体的部分端粒缺失;②Ohdo型,只局限于Ohdo报道的原生家庭的患者;③SBBYS型,具有典型畸形特征,包括心脏缺损、视神经萎缩、耳聋、牙齿发育不良、唇腭裂、关节受限和甲状腺功能减退症。SBBYS类型可能是一种病因异质性表型,常染色体显性遗传和常染色体隐性遗传的形式可同时存在;④BMRS 的MKB(Maat Kievit Brunner)表型,表现为面容粗糙、三角脸,以男婴多见,可能与性别相关;⑤BMRS的V(verloes)表型,可能是一个新类型的严重畸形,包括严重心律失常,拇指内收,腭裂和生殖器异常,属于常染色体隐性遗传。2008年Day等[16]又将SBBYS型更新分为3型,分别为 1型、Ohdo-Like综合征(OLS)和Young-Simpson综合征(YSS)。还可见患儿存在隐匿的染色体重排情况的报道,从而进一步扩大了BMR的疾病谱[17,18]。由此可见,BMR分型众多,临床特点存在些许差异。

图2UBE3B基因c.2737C>T (exon25)杂合位点突变

图3 患儿照片

图4 患儿X线胸片
本例患儿通过基因检测,明确诊断为由于UBE3B基因的复合杂合突变导致的BPID。BPID是一种罕见的常染色体隐性遗传病,在中国知网和万方数据库中未查到相关报道,在PubMed数据库中检索,仅找到关于BPID的报道1篇文献,由Basel-Vanagaite等[2]首次报道于2012年,共来自3个不同家庭的4例患儿,加本例患儿,目前仅5例,均为UBE3B基因突变导致,属于BMR的一种新类型。通过复习和总结BPID的文献和病例,将患儿的临床特点和影像、生化、基因等进行比较,绘制成表格,见表1,主要摘译自Basel-Vanagaite等的报道[2]。
BPID患儿共同的临床特点包括:①围生期存在呼吸疾病、喂养困难、生长迟缓等;②存在小头畸形、睑裂狭小、内眦距过宽、耳位低/发育异常、小下颌、小嘴等面部畸形;③运动、智力和语言发育存在异常或严重异常;④部分病例还存在眉毛浅淡、皮肤薄等皮肤特征,泌尿生殖畸形,心脏畸形,胼胝体发育异常以及视听功能损害。其中3例患儿还表现为血清胆固醇降低,提示胆固醇代谢缺陷可能是BPID的部分表现。因本例患儿未作相关检测,且年龄尚小,需要进一步随访证实。5例患儿均通过外显子组测序检查,确定了UBE3B基因突变致病,属于常染色体隐性遗传病。因UBE3B基因主要负责编码泛素连接酶E3B,泛素/蛋白酶体系统降解受损的蛋白质,是细胞蛋白质控制网络的一部分,而泛素化是一种蛋白质翻译后的修饰,在大脑发育中起关键作用[19-21],有助于调节神经元迁移,轴突和突触的形成和消除,因此泛素/蛋白酶体系统异常改变可以导致神经功能障碍,造成神经系统疾病和智力低下[20]。目前外显子基因测序技术的推广应用增加了单基因疾病和新的疾病相关基因的检出率[22],为今后精准医学在优生优育国策中的应用创造了良好条件。

表1 5例BPID患儿临床特征
BMR是在20世纪80年代首次被描述[1],是一组由不同基因突变导致的包含多重先天性畸形的综合征,包括一组临床和病因异质性的表型,可作为孤立的特征或多个先天性异常的不同疾病的一部分出现。BMR分型众多,本例患儿所属的BPID是BMR一个新的罕见分型,其主要临床特征包括面部畸形,喂养困难,喉软骨软化,生长发育迟缓,运动、智力发育异常,以及胼胝体发育不良,泌尿生殖畸形、心脏畸形、视听损害等表现。本例患儿通过全外显子基因测序技术,确诊为UBE3B基因复合杂合突变导致的BPID,其突变位点之一在OMIM数据库和dbSNP数据库中未见报道,可能是UBE3B基因新的致病突变位点。由于本病预后不佳,患儿生存质量差,如果患儿父母希望再次妊娠,需进行遗传咨询,并于产前行羊水穿刺,将胎儿脱落细胞基因与父母的基因进行比对,指导妊娠。
通过本例患儿及文献复习,提示在临床中发现睑裂狭小、上睑下垂、小下颌等面部畸形,伴有喉软骨软化,生长发育迟缓,胼胝体发育不良,泌尿生殖畸形、心脏畸形等表现的患儿,需要考虑本病可能性。由于本病可引起生长发育、语言、运动和智力以及视听等多方面的落后和损害,患儿生存能力差,且预后不佳,确诊主要依靠基因检测,无有效治疗方法,因此高危家庭进行遗传咨询和指导尤为重要。
[1]Ohdo S, Madokoro H, Sonoda T, et al. Mental retardation associated with congenital heart disease, blepharophimosis,blepharoptosis, and hypoplastic teeth [J]. J Med Genet, 1986,23(3): 242-244.
[2]Basel-Vanagaite L, Dallapiccola B, Ramirez-Solis R,et al. Deficiency for the ubiquitin ligaseUBE3Bin a Blepharophimosis-Ptosis-Intellectual-Disability syndrome [J].Am J Hum Genet, 2012, 91(6):998-1010.
[3]Marcellus L. The infant with Pierre Robin sequence:review and implications for nursiIlg practice [J]. J Pediatr Nuns,2001, 16(1):23-24.
[4]Wigren M, Hansen S. Prader-Willi syndrome: clinical picture,psychosocial support and current management [J]. Child Care Health Dev, 2003, 29(6): 449-456.
[5]Guercio JR, Martyn LJ. Congenital malformations of the eye and orbit [J]. Otolaryngol Clin North Am, 2007, 40(1): 113-140.
[6]Hall BD, Graham JM Jr, Cassidy SB, et al. Elements of morphology: standard terminology for the periorbital region[J]. Am J Med Genet Part A, 2009, 149A(1): 29-39.
[7]Zlotogora J, Sagi M, Cohen T. The blepharophimosis, ptosis,and epicanthus inversus syndrome: delineation of two types[J]. Am J Hum Genet, 1983, 35(5):1020-1027.
[8]Dubowitz V. Familial low birth weight dwarfism with an unusual facies and a skin eruption [J]. J Med Genet, 1965,2(1):12-17.
[9]Marden PM, Walker WA. A new generalized connective tissue syndrome [J]. Am J Dis Child, 1966, 112(3):225-228.
[10]Say B, Barber N. Mental retardation with blepharophimosis[J]. J Med Genet, 1987, 24(8): 511-511.
[11]Young ID, Simpson K. Unknown syndrome: abnormal facies, congenital heart defects, hypothyroidism, and severe retardation [J]. J Med Genet, 1987, 24(11):715-716.
[12]Clayton-Smith J, Krajewska-Walasek M, Fryer A, et al.Ohdo-likeblepharophimosis syndrome with distinctive facies,neonatal hypotonia, mental retardation and hypoplastic teeth[J]. Clin Dysmorphol, 1994, 3(2):115-120.
[13]Biesecker LG. The Ohdo blepharophimosis syndrome: a third case [J]. J Med Genet, 1991, 28(2): 131-134.
[14]Melnyk AR. Blepharophimosis, ptosis and mental retardation:further delineation of Ohdo Syndrome [J]. Clin Dysmorphol,1994, 3(2):121-124.
[15]Verloes A, Bremond-Gignac D, Isidor B, et al. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdosyndrome, and delineation of two new BMR syndromes,one X-linked and one autosomal recessive [J]. Am J Med Genet A, 2006,140(12):1285-1296.
[16]Day R, Beckett B, Donnai, et al. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdosyndrome [J]. Clin Genet, 2008, 74(5): 434-444.
[17]Bartholdi D, Toelle SP, Steiner B, et al. Blepharophimosis and mental retardation (BMR) phenotypes caused by chromosomal rearrangements: description in a boy with partial trisomy 10q and monosomy 4q and review of the literature [J]. Eur J Med Genet, 2008, 51(2): 113-123.
[18]Brancati F, Bernardini L, Cavalcanti DP, et al. Genome rearrangements in patients with blepharophimosis, mental retardation and hypothyroidism, so-called Young-Simpson syndrome [J]. Clin Genet, 2009, 76(2):210-213.
[19]Kawabe H, Brose N. The role of ubiquitylation in nerve cell development [J]. Nat Rev Neurosci, 2011, 12(5): 251-268.
[20]Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction [J]. Nat Rev Neurosci, 2008, 9(11):826-838.
[21]Komander D. The emerging complexity of protein ubiquitination [J]. Biochem Soc Trans, 2009, 37(5):937-953.
[22]Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery [J]. Nat Rev Genet, 2011, 12(11):745-755.
《临床儿科杂志》征订启事
《临床儿科杂志》创刊于1983年,由上海市儿科医学研究所及上海交通大学医学院附属新华医院主办,编委会由近200名全国各地的著名儿科专家、教授组成。本刊遵循面向临床、面向基层、普及与提高相结合的办刊宗旨,反映儿科领域学术水平和发展动向。主要读者为二、三级医院的医务人员,每期除设1个系统疾病专栏(包括呼吸、消化、神经、血液、心血管、肾脏、免疫、遗传、代谢、内分泌等系统)外,另设综合报道、实验研究、文献综述、临床经验点滴和继续医学教育讲座等栏目。本刊自20世纪90年代起连续被评为临床医学类及生物医学类核心期刊,2000年起被纳入国家科学技术部中国科技论文统计源期刊,且成为中国科学引文数据库来源期刊,并被国际著名检索机构美国《化学文摘》、波兰《哥白尼索引》收录。
本刊为月刊,每月15日出版,公开发行,2017年每期定价15元,全年定价180元。国内报刊邮发代号:4-426,国外发行代号M5788。编辑部地址:上海市杨浦区控江路1665号;邮编:200092;电话:(021)25076489;网站:http://www.jcp-sh.org.cn。
热诚欢迎新、老读者订阅。
Blepharophimosis-ptosis-intellectual-disability syndrome: a case report and literature review
CAO Lifang1, TONG Xiaomei2, TIAN Yaping1, SONG Lin1(1. Department of Pediatrics, Peking University International Hospital, Bejing 102206,China; 2. Department of Pediatrics, Peking University Third Hospital, Bejing 100191, China)
ObjectiveTo explore the clinical features and gene mutations of blepharophimosis-ptosis-intellectualdisability syndrome (BPID).MethodsThe clinical data, diagnosis and treatment of a child with BPID in neonatal intensive care unit (NICU) were reviewed. Based on the literature retrieved from PubMed database, the common classifcation, clinical features, diagnosis and genetic counseling of BPID and its affliated blepharophimosis-mental retardation syndromes (BMR) were reviewed.ResultsThis male infant was 39 weeks of gestational age with birth weight of 1920 g, and was admitted to NICU 15 min after birth due to dyspnea. The main clinical manifestations were facial deformity such as biepharophimosis, ptosis and micromandible, inspiratory dyspnea with laryngeal cartilage softening, malformations of the thorax and feeding diffculties. A heterozygous mutation inUBE3Bgene was identifed by complete exon sequencing and he was diagnosed of BPID, a rare genetic disorder. Reviewing the literature, there was no relevant report in domestic. While one foreign literature was found to report 5 patients from 4 families having a subtype of BMR, a kind of autosomal recessive diseases caused by mutations in theUBE3Bgene. Conclusion BPID is a rare clinical entity of BMR. Complete exon sequencing can be used to diagnose the disease.
biepharophimosis; ptosis; mental retardation; gene mutation
2017-03-10)(本文编辑:邹 强)
10.3969/j.issn.1000-3606.2017.07.018
童笑梅 电子信箱:tongxm2007@126.com
