BSCL2基因突变致先天性全身脂肪营养不良症1例报告并文献复习
2017-07-31张梦奇马明圣邱正庆
张梦奇 马明圣 邱正庆
中国医学科学院 北京协和医学院 北京协和医院儿科(北京 100730)
BSCL2基因突变致先天性全身脂肪营养不良症1例报告并文献复习
张梦奇 马明圣 邱正庆
中国医学科学院 北京协和医学院 北京协和医院儿科(北京 100730)
目的探讨先天性全身脂肪营养不良症(CGL)的临床及基因特点。方法回顾分析1例BSCL2基因突变致CGL患儿的临床资料,并进行文献复习。结果女性患儿,2岁9个月,临床表现为全身脂肪组织消失,黑棘皮征,肝脾大,轻度智力低下;实验室检查示高三酰甘油血症、高胰岛素血症和心肌病变。提取患儿及父母外周血,对AGPAT2、BSCL2、CAV1和PTRF 4个基因行Sanger测序显示,患儿存在BSCL2基因杂合突变,分别为母源移码突变(c.567-568delGA,p.E189EfsX12)及父源无义突变(c.565G>T,p.E189X),均为致病突变。回顾文献,BSCL2基因突变是亚洲CGL最常见的病因,BSCL2突变的CGL患儿常见临床表现为全身脂肪组织消失、黑棘皮征和肝脾大,心肌病变和智力低下发生率分别为40%和30%。结论BSCL2基因突变引起的CGL主要临床表现为自幼全身脂肪组织消失及代谢紊乱,常伴有心肌病变和智力低下,对疑似患儿应尽早行基因分析确诊。
先天性全身脂肪营养不良;BSCL2基因; 杂合突变
先天性全身脂肪营养不良症(congenial generalized lipodystrophy,CGL)又称Berardinelli-Seip综合征(Berardinelli-Seip congenial lipodystrophy),是一种罕见的常染色体隐性遗传病,在新生儿中的发病率约为1/12 000。临床表现为全身脂肪组织消失,可继发糖代谢和脂代谢异常,产生严重的并发症如糖尿病、高三酰甘油血症、脂肪肝、肝硬化,预后不佳[1]。目前已知的CGL相关基因共4个,其中BSCL2基因突变(CGL2型)是最为常见的类型,约占报道病例的90%[2]。自1954年首次报道以来,全球各地陆续报到,多为非亚洲病例,而国内文献基因诊断明确病例少见。现报告1例临床和基因诊断均明确的CGL患儿,并进行文献复习。
1 临床资料
患儿,女,2岁9个月,山西人。因发现脂肪菲薄、肝脾大2年余就诊于北京协和医院儿科。患儿生后3个月时家长发现其全身消瘦、皮肤弹性差,无腹泻、呕吐,无发热,无皮疹,无多尿,吃奶可。生后6个月因“消瘦”于当地儿童医院住院,查血常规、肝肾功能、血三酰甘油水平均正常。血涂片及骨髓检查未见异常。腹部B超:肝肋下4.1 cm,右叶最大斜径7.4 cm,实质回声不均;脾大,肋上厚2.4 cm,肋下2.4 cm。颅脑、垂体磁共振成像(MRI)未见明确异常。染色体核型为“46,XX”。尿、血代谢病筛查:草酸、3羟基丙酸、甘油酸、3羟基苯乙酸浓度增高,提示营养障碍可能。尿液分析未见典型有机酸代谢病改变。当地诊断“营养不良、肝脾大待查,遗传代谢病可能性大”,未予特殊治疗。患儿自1岁后食欲旺盛,肤色加深。患儿系G3P1,母亲孕期有保胎史。足月顺产,出生体质量2.3 kg,身长50 cm,无窒息抢救史。2个月抬头,6个月独坐,10个月翻身,1岁独站,1岁半独走,2岁半跑步,1岁说单音、1岁余叫“爸爸、妈妈”,目前说若干单字,无法连贯成句。入院时Gesell发育评估:语言行为、个人-社交行适应性行为落后(具体不详)。父母体健,非近亲结婚。家族中无类似疾病患者。
入院体格检查:体质量 16 kg,身高101 cm;神志清;皮肤色深,颈部、腋下、肘窝、腹股沟、腘窝等皮肤皱褶处色素沉着,头发稍卷曲,浓密,四肢及背部毛发较多,全身皮下脂肪菲薄;下颌骨突出,耳大;肺部查体未见异常;胸骨左缘第三肋间可闻及Ⅲ/6级收缩期吹风样杂音;腹膨隆,肝右肋下4 cm,剑下4 cm,质韧,表面光滑,脾肋下及边;脊柱四肢无畸形,四肢肌肉发达,血管显露;幼女型外阴,阴蒂肥厚。实验室检查:丙氨酸转移酶(ALT )76U/L,天冬氨酸转移酶(AST) 42 U/L,谷氨酰转肽酶(GGT )95 U/L,白蛋白 (Alb) 51 g/L,三酰甘油(TG) 3.02 mmol/L,胆固醇(TC )4.32 mmol/L,高密度脂蛋白胆固醇(HDL) 0.65 mmol/L,低密度脂蛋白胆固醇(LDL)2.83 mmol/L;血电解质正常;糖化血红蛋白4.0%,空腹血糖82 mg/dL,餐后2小时血糖92 mg/dL;空腹胰岛素44.89μlU/mL、C-肽 5.29 ng/mL,餐后2小时胰岛素115.60μlU/mL、C-肽10.21 ng/mL。8am促肾上腺素皮质激素(ACTH)、皮质醇、17羟孕酮未见异常。腹部B超:肝剑下6.3 cm,肋下4.3 cm,右叶斜径12.2 cm,肝回声均匀;脾厚3.4cm,长径11.5 cm,肋下1.6 cm。腹部CT平扫:肝脾增大,重度脂肪肝。全身弥散加权成像提示颌面部、腰背部、双侧髂前上棘前方、双侧臀部、双上臂后内侧、双前臂及双下肢皮下脂肪菲薄。颈动脉、椎动脉超声:双侧椎动脉阻力指数增高,双侧颈动脉外膜回声稍强。心电图大致正常。超声心动图:左室肥厚,主动脉瓣右冠瓣回声增强,主动脉瓣轻度关闭不全。骨龄:相当于3.5岁左右。垂体MRI、肾上腺B超、子宫双附件B超未见明显异常。
经医院伦理委员会审核,父母知情同意情况下,抽取患儿及其父母静脉血各2 mL,采用血液基因组DNA提取试剂盒提取白细胞DNA,用eppendrof公司紫外分光光度计检测DNA纯度,纯度经鉴定合格的样本放入-80℃冰箱保存待用。根据UCSC数据库基因序列,应用Primer3软件在线设计引物,使用ABI9700PCR仪进行PCR扩增AGPAT2、BSCL2、CAV1、PTRF基因全部编码区及其旁侧内含子区域,DNA自动分析仪检测扩增产物。患儿BSCL2基因发现2个突变位点:c.567_568delGA(对应氨基酸改变为p.E189EfsX12,其母携带有该位点的杂合突变)和c.565G>T(对应氨基酸改变为p.E189X,其父携带有该位点的杂合突变),见图1。此两种突变类型均可导致其编码的蛋白质发生截断突变,致病性明确。

图1BSCL2基因突变位点Sanger测序图
2 讨论
自1954年Berardinelli首次报道CGL病例以来,国内外陆续有报道。利用Pubmed、OVID及Elsevier和国内CNKI、万方医学文献数据库,用先天性全身脂肪营养不良、脂肪萎缩性糖尿病、BSCL2、congenital generalized lipodystrophy为关键词进行检索,得到2000年1月-2016年10月之间亚洲范围内临床诊断和BSCL2基因突变诊断明确的英文CGL完整病例报道12篇[3-13],共18例,连同本例共19例CGL中BSCL2基因突变患者(表1)。国内数据库检索13例,基因诊断明确者3例,均为BSCL2基因突变所致。
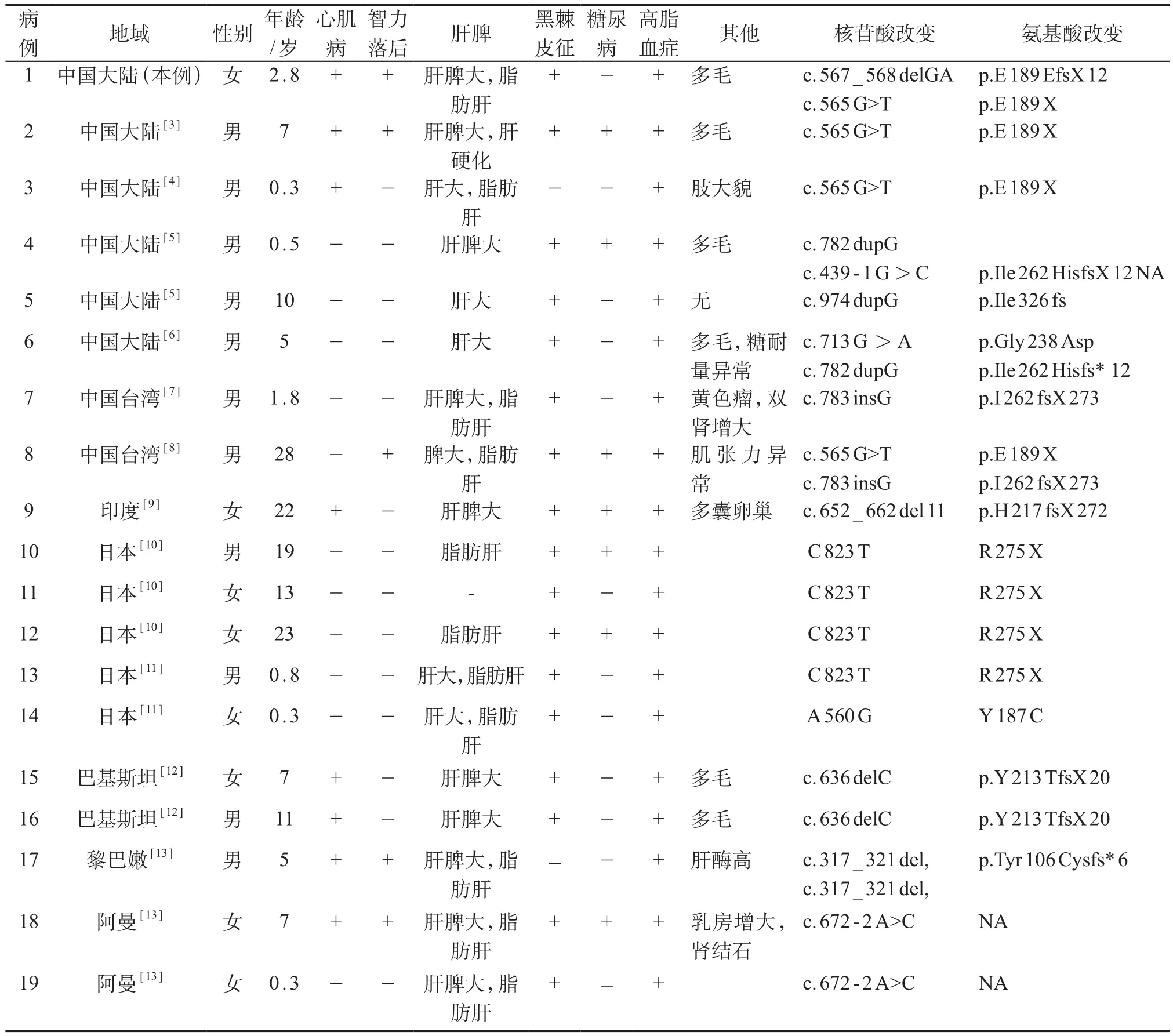
表1 19例CGL患儿临床及基因特点
19例CGL中BSCL2基因突变患者种族背景各异,其中男11例,女8例。起病年龄在出生后7月内,7例(37%)患儿的基因诊断年龄在3岁以内,诊断时间延迟多与基因检测技术限制以及既往对于该病认识有限相关。全身脂肪消失是最常见的临床表现,19例患儿均有,其他临床表现和体征有黑棘皮征17例,肝大14例,脾大11例,智力落后5例。实验室检查异常以高三酰甘油血症最为常见,19例均有,其他异常包括脂肪肝12例,高血糖7例,心肌病8例等。糖尿病多于10~15岁起病,最小1例糖尿病于新生儿期起病。
CGL主要临床表现为生后不久出现的几乎全部的脂肪组织缺失,缺乏脂肪组织可导致严重的糖代谢和脂代谢异常,脂肪异位沉积于肝脏导致肝大、脂肪肝、胰岛素抵抗和糖尿病。患儿在婴幼儿时期可出现肝脾大、脐疝;幼年期可出现黑棘皮征,食欲旺盛、生长加速;儿童期时女童可有多毛、阴蒂肥大;青春期女孩有月经不规律等多囊卵巢的表现[1]。2015年提出CGL诊断标准:出生或者生后不久出现的几乎全部脂肪组织消失,肌肉显露,查体可见特征性脂肪分布或通过MRI进行分型,并通过基因检测明确诊断[14]。本例患儿生后3个月即出现特殊面容、全身消瘦、肌肉血管显露、肝脾肿大,入院查体及全身MRI检查均提示全部脂肪组织消失,肝脏脂肪异位沉积,黑棘皮征,血三酰甘油升高,临床拟诊CGL,行CGL相关基因检测明确诊断。
目前已知的CGL相关基因有4个,分别是位于染色体9q34的AGPAT2(1-acylglycerol-3-phosphate O-acyltransferase 2)、位于染色体11q13的BSCL2(Berardinelli-Seip congenital lipodystrophy 2)、CAV1(caveolin 1)、PTRF(polymerase I and transcript release factor)。绝大多数的CGL病例由AGPAT2或BSCL2突变导致。BSCL2编码的seipin蛋白,参与脂滴的形成和脂肪细胞的分化,且该基因突变可破坏蛋白的N-糖基化位点,导致蛋白质的错误折叠及神经元变性[15]。BSCL2突变导致的CGL表型最重,表现为生后不久即有全部脂肪组织消失,包括参与代谢的脂肪组织和保护性脂肪组织,并可伴随神经系统表现和心肌病变。其中,神经系统表现包括轻度的智力发育落后、肌张力异常、共济失调性步态及癫痫[16]。BSCL2基因突变亦与远端遗传性运动神经病5型、Silver综合征、Charcot-Marie-Tooth 病2型等神经系统疾病相关,这些疾病临床共同点包括:起病年龄不一、进展缓慢、上运动神经元及下运动神经元受累、感觉大多正常、弓形足等足部畸形。本例患儿有CGL的典型临床表现,无神经元受累表现,基因结果诊断明确。
BSCL2基因突变呈现多样化。截至2013年共发现31种突变中,错义或者无义突变12种,微小插入4种,微小缺失4种,剪切位点突变7种,复合重组4种[12]。由此可见,错义或者无义突变最为常见。BSCL2基因突变所致CGL报道在我国并不多见,总结的7例中基因突变亦呈现多样化,以错义及微小插入为主。本例患儿为复合杂合突变,均位于第6外显子,引起蛋白质截断突变,其中c.567_568delGA目前未见文献报道。
CGL临床上应注意和有全身脂肪萎缩的疾病相鉴别,如获得性全身性脂肪营养不良(acquired generalized lipodystrophy,AGL)、早老症(neonatal progeroid syndrome,OMIM: 176670)。AGL与自身免疫或者感染相关,青春期或成人早期起病,女性多见,临床亦可表现为全身脂肪组织消失、高三酰甘油、高胰岛素血症和糖尿病,可通过发病年龄、病因查找及基因诊断鉴别。早老症为罕见常染色隐性遗传病,多见于Lamin A/C基因突变导致,亦可有全身皮下脂肪消失,三酰甘油增高,但缺少多毛、四肢肌肉发达和高代谢状态等临床表现。另外,BSCL2基因突变所致CGL应与CGL其他亚型相鉴别。BSCL2基因突变为CGL最常见病因,其次为AGPAT2基因突变。AGPAT2基因所致CGL主要临床特点为生后不久出现的皮下等参与代谢的脂肪组织消失,而眶周、掌心、关节周围等的保护性脂肪组织尚存。本例患儿参与代谢及保护性脂肪组织均消失,符合BSCL2基因突变所致CGL临床表现。
CGL目前尚无特效治疗方法。疾病初期饮食控制是最重要的治疗手段。低脂饮食、限制饱和脂肪酸摄入、适当限制高糖和高热量食物的摄入有助于控制血脂。对于胰岛素抵抗性糖尿病,二甲双胍为首选用药,因其同时能够抑制食欲,改善多囊卵巢和脂肪肝的症状[17]。一项对于9名CGL和瘦素缺乏患者给予1年瘦素治疗的研究表明,瘦素能够显著降低三酰甘油水平,改善胰岛素抵抗,并抑制食欲、降低空腹血糖和糖化血红蛋白水平[18]。糖尿病、高脂血症和脂肪肝是CGL治疗的难点。CGL预后不佳,糖尿病肾病、糖尿病视网膜病变、反复发作的胰腺炎和肝硬化食道静脉曲张破裂出血为主要的死因。
综上,CGL主要表现为生后不久出现的全身脂肪组织消失,逐渐出现的代谢紊乱。BSCL2基因突变引起的先天性全身脂肪营养不良常伴随心肌病变及智力地下。对疑似患儿应尽早行基因分析确诊。
[1]Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome [J]. Endocrinol Metab Clin North Am,2004, 33(2): 305-331.
[2]Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders [J]. J Clin Endocrinol Metab,2011, 96(11): 3313-3325.
[3]Jin J, Cao L, Zhao Z, et al. NovelBSCL2gene mutation E189X in Chinese congenital generalized lipodystrophy child with early onset diabetes mellitus [J]. Eur J Endocrinol, 2007,157(6): 783-787.
[4]Friguls B, Coroleu W, del Alcazar R, et al. Severe cardiac phenotype of Berardinelli-Seip congenital lipodystrophy in an infant with homozygous E189XBSCL2mutation [J]. Eur J Med Genet, 2009, 52(1): 14-16.
[5]劳文芹,孟哲,欧辉,等. 儿童Berardinelli-Seip综合征2例报告[J]. 中国实用儿科杂志, 2016, 2: 157-158.
[6]袁欣,陈瑞敏,王剑,等. 先天性全身性脂肪营养不良BSCL2基因突变1例并文献复习[J]. 中国循证儿科杂志,2016, (5): 377-381.
[7]Huang HH, Chen TH, Hsiao HP, et al. A Taiwanese boy with congenital generalized lipodystrophy caused by homozygous Ile262fs mutation in theBSCL2gene [J]. Kaohsiung J Med Sci, 2010, 26(11): 615-620.
[8]Wu YR, Hung SI, Chang YC, et al. Complementary mutations in seipin gene in a patient with Berardinelli-Seip congenital lipodystrophy and dystonia: phenotype variability suggests multiple roles of seipin gene [J]. J Neurol Neurosurg Psychiatry, 2009, 80(10): 1180-1181.
[9]Shirwalkar HU, Patel ZM, Magre J, et al. Congenital generalized lipodystrophy in an Indian patient with a novel mutation inBSCL2gene [J]. J Inherit Metab Dis, 2008, 31Suppl 2: s317-s322.
[10]Ebihara K, Kusakabe T, Masuzaki H, et al. Gene and phenotype analysis of congenital generalized lipodystrophy in Japanese: a novel homozygous nonsense mutation in seipin gene [J]. J Clin Endocrinol Metab, 2004, 89(5): 2360-2364.
[11]Nishiyama A, Yagi M, Awano H, et al. Two Japanese infants with congenital generalized lipodystrophy due toBSCL2mutations [J]. Pediatr Int, 2009, 51(6): 775-779.
[12]Rahman OU, Khawar N, Khan MA, et al. Deletion mutation inBSCL2gene underlies congenital generalized lipodystrophy in a Pakistani family [J]. Diagn Pathol, 2013, 8:78.
[13]Haghighi A, Kavehmanesh Z, Haghighi A, et al. Congenital generalized lipodystrophy: identification of novel variants and expansion of clinical spectrum [J]. Clin Genet, 2015, doi:10.1111/cge.12623.
[14]Patni N, Garg A. Congenital generalized lipodystrophies-new insights into metabolic dysfunction [J]. Nat Rev Endocrinol,2015, 11(9): 522-534
[15]Windpassinger C, Auer-Grumbach M, Irobi J, et al.Heterozygous missense mutations inBSCL2are associated with distal hereditary motor neuropathy and Silver syndrome[J]. Nat Genet, 2004, 36(3): 271-276.
[16]Opri R, Fabrizi GM, Cantalupo G, et al. Progressive myoclonus epilepsy in congenital generalized lipodystrophy type 2: Report of 3 cases and literature review [J]. Seizure, 2016, 42: 1-6.
[17]Garg A. Acquired and inherited lipodystrophies [J]. N Engl J Med, 2004, 350(12): 1220-1234.
[18]Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy [J]. N Engl J Med, 2002, 346(8): 570-578.
Congenital generalized lipodystrophy caused by mutation ofBSCL2gene: a case report and literature review
ZHANG Mengqi, MA Mingsheng, QIU Zhengqing (Department of Pediatrics, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China)
ObjectiveTo explore the clinical and genetic characteristics of congenital generalized lipodystrophy (CGL).MethodThe clinical data of one child with CGL caused byBSCL2gene mutation were analyzed retrospectively and relative literature were reviewed.ResultsA 2-year-9-month old girl had clinical manifestations of a lack of subcutaneous fat, acanthosis nigricans, hepatolienomegaly and mild hypophrenia. Laboratory examinations showed hypertriglyceridemia, hyperinsulinemia and cardiomyopathy. The peripheral blood from the child and her parents were collected and 4 genes,AGPAT2,BSCL2,CAV1andPTRF, were sequenced by Sanger. The results showed a heterozygous mutation ofBSCL2gene from maternal frameshift (c.567-568delGA, p.E189EfsX12) and paternal nonsense mutation (c.565G>T,p.E189X) respectively in the child, and both mutations were pathogenic ones. By a literature review, it is known thatBSCL2gene mutation is the most common cause of in Asian. In CGL withBSCL2gene mutation, the commom clinical manifestations include disappearance of systemic adipose tissue, acathosis nigricans and hepatomegaly, and the incidence of myocardial infarction and mental retardation were 40% and 30% respectively.ConclusionThe main clinical manifestations of CGL caused byBSCL2gene mutation were loss of systemic adipose tissue and metabolic disorder at an early age. It was often accompanied by myocardial lesions and mental retardation. Gene diagnosis analysis should be made as earliest possible time for the children suspected of this disease.
congenial generalized lipodystrophy;BSCL2gene; heterozygous mutation
2016-12-26)
(本文编辑: 梁 华)
10.3969/j.issn.1000-3606.2017.07.015
邱正庆 电子信箱:zhengqingqiu33@aliyun.com
