DHA通过抑制PI3K通路和GLUT2表达而诱导NSCLC细胞毒性
2016-01-12闵发胜,杨建洪,黄建军等
DHA通过抑制PI3K通路和GLUT2表达而诱导NSCLC细胞毒性
闵发胜1,杨建洪1,黄建军1,刘启梁2△
(1国防科技大学医院保健科,湖南 长沙 410073;2第三军医大学病理生理学教研室,重庆 400037)
[摘要]目的: 研究二氢青蒿素(dihydroartemisinin, DHA)诱导NSCLC细胞毒性的分子机制。方法: 将不同浓度的DHA作用NSCLC细胞系A549和NCI-H1650细胞不同时间,运用MTT法、克隆形成实验、Annexin V染色和流式细胞术测定DHA对细胞活力、克隆形成能力和细胞凋亡的影响;同时测定DHA对细胞中葡萄糖水平、ATP和乳酸含量的影响;Western blot检测PI3K通路活性和GLUT2表达变化;通过细胞转染实现葡萄糖转运体2(GLUT2)和Rheb在A549和NCI-H1650细胞中高表达,测定和分析DHA作用下细胞活力、细胞凋亡、葡萄糖水平、ATP含量和PI3K通路活性的变化;分析葡萄糖缺乏对DHA诱导NSCLC细胞毒性的影响。结果: 与对照组比较,DHA显著抑制A549和NCI-H1650细胞活力和克隆形成能力以及诱导细胞凋亡,同时降低ATP和乳酸含量以及抑制细胞对葡萄糖的摄取,具有时间和剂量依赖效应。Western blot结果显示,DHA能抑制PI3K通路活性和GLUT2的表达。上调GLUT2的表达和激活PI3K通路能减弱DHA对NSCLC细胞的毒性作用;葡萄糖缺乏能增强DHA对NSCLC细胞的毒性;相反,高浓度葡萄糖则抑制DHA对NSCLC细胞的毒性。结论: DHA能抑制NSCLC细胞活力和克隆形成,诱导细胞凋亡,该作用是通过降低PI3K活性和GLUT2的表达进而抑制细胞糖酵解代谢实现的。
[关键词]非小细胞肺癌; 二氢青蒿素; PI3K通路; 葡萄糖转运体2; 细胞毒性
[中图分类号]R730.23[文献标志码]A
doi:10.3969/j.issn.1000-4718.2015.11.007
[文章编号]1000-4718(2015)11-1970-09
[收稿日期]2015-05-11[修回日期] 2015-06-17
通讯作者△Tel: 0719-8272465; E-mail: Zhengxin19740912@163.com
DHA induced cytotoxicity in NSCLC cells via inhibiting PI3K pathway activation and GLUT2 expressionMIN Fa-sheng1, YANG Jian-hong1, HUANG Jian-jun1, LIU Qi-liang2
(1DepartmentofHealthCare,HospitalofNationalUniversityofDefenseTechnology,Changsha410073,China;2DepartmentofPathophysiology,ThirdMilitaryMedicalUniversity,Chongqing400037,China.E-mail:zhongliukeyan@163.com)
ABSTRACT[]AIM: To investigate the molecular mechanisms of cytotoxicity induced by dihydroartemisinin (DHA) in non-small cell lung cancer (NSCLC) cells. METHODS: NSCLC cell lines A549 and NCI-H1650 were treated with various concentrations of DHA for indicated time. Subsequently, the effects of DHA on the cell activity, colony formation ability and apoptosis were determined by MTT assay, colony formation assay, Annexin V-FITC/PI staining and flow cytometry, respectively. At the same time, the effects of DHA on glucose, ATP and lactate levels were assessed, and the PI3K pathway activation and glucose transporter 2 (GLUT2) expression were detected by Western blot in the A549 cells and NCI-H1650 cells. Overexpression of GLUT2 and Rheb was established in A549 and NCI-H1650 cells by transfection with GST-GLUT2 and GST-Rheb plasmids, respectively, and the effects of DHA on cell activity, apoptosis, glucose level, ATP content and PI3K pathway activation were analyzed in A549 cells and NCI-H1650 cells. The effect of glucose deprivation on the cytotoxicity triggered by DHA in NSCLC cells was also determined. RESULTS: Compared with control group, DHA significantly inhibited cell activity and colony formation ability, and induced remarkable cell apoptosis in the A549 cells and NCI-H1650 cells. At the same time, DHA reduced ATP and lactate contents, and hindered glucose uptake in a time- and dose-dependent manner in A549 cells and NCI-H1650 cells. The activity of PI3K pathway and GLUT2 expression were downregulated, while upregulated GLUT2 expression and activated PI3K pathway reduced the cytotoxicity induced by DHA in NSCLC cells. Glucose deprivation increased DHA-mediated cytotoxicity in NSCLC cells. On the contrary, high levels of glucose inhibited DHA-mediated cytotoxicity in NSCLC cells. CONCLUSION: DHA restrains cell activity and colony formation, and induces apoptosis. DHA induces cytotoxicity via inhibiting PI3K pathway activation and GLUT2 expression, leading to inhibit glycolytic metabolism in NSCLC cells.
[KEY WORDS]Non-small cell lung cancer; Dihydroartemisinin; PI3K pathway; Glucose transporter 2; Cytotoxicity
肺癌是当今世界上严重威胁人类生命与健康的最常见的恶性肿瘤之一,其发病率和死亡率在我国居第3位。非小细胞肺癌(non-small cell lung can-cer,NSCLC)是肺癌的主要类型,NSCLC对一些抗癌药物如它莫西芬和芳香酶抑制剂不敏感[1]。因此,寻找和研发靶向治疗NSCLC的新型药物是当前亟需解决的问题。二氢青蒿素(dihydroartemisinin,DHA)是一种天然存在于青蒿素中具有抗疟疾功能的中草药[2]。最新的研究发现[3-4],DHA能抑制乳腺癌、宫颈癌、肝癌和胰腺癌细胞增殖和诱导细胞凋亡,提示DHA具有抗肿瘤功能。同时有资料显示[5],DHA通过诱导细胞凋亡引起NSCLC细胞毒性,而对正常细胞无影响。另研究表明DHA对肿瘤细胞增殖、血管生成和免疫调节具有重要的调控作用[6-8]。然而,DHA抗肿瘤功能的分子机制目前仍不完全清楚。
癌细胞内的生物合成速度和葡萄糖代谢反应加快,使得癌细胞经常遭遇代谢应激和营养匮乏[9]。因此,癌细胞通过促进葡萄糖摄取和糖酵解代谢从而缓解肿瘤细胞对营养物的需求[10]。激活mTOR/PI3K通路促进细胞蛋白质合成和细胞增殖,同时PI3K通路在细胞糖代谢过程中发挥着重要的调控作用[11]。最新的研究表明激活mTOR通路能够促进肿瘤细胞对葡萄糖的摄入从而促进葡萄糖代谢[12]。葡萄糖转运体(glucose transporter, GLUT)是细胞摄取葡萄糖所必须,研究发现GLUT在大多数肿瘤细胞中高表达,而在正常上皮细胞中不表达[13-14]。促进GLUT的表达能够加速细胞对葡萄糖转运从而缓解肿瘤细胞对葡萄糖的需求[10]。早期的研究发现雷帕霉素抑制mTOR通路活性进而抑制GLUT1的表达最终抑制细胞对葡萄糖的摄入[15]。目前,DHA诱导NSCLC细胞毒性作用的分子机制仍不清楚。因此,本研究以非小细胞肺癌细胞系A549和NCI-H1650细胞为对象,研究DHA诱导NSCLC细胞毒性作用可能的分子机制。
材料和方法
1细胞株和材料
非小细胞肺癌细胞株A549和NCI-H1650细胞购自中国科学院上海细胞生物研究所;ATP生物发光检测试剂盒、氧化酶法-葡萄糖测定试剂盒、乳酸测定试剂盒、MTT、DMSO、葡萄糖、DHA购自Sigma;DHA溶解于DMSO中,-20℃保存,作用细胞前用DMEM培养液稀释至所需浓度;DMEM培养基(Gibco);胎牛血清(fetal bovine serum,FBS)、胰蛋白酶和双抗购自北京金博益生物技术有限公司;Annexin V-FITC/PI双染细胞凋亡检测试剂盒购自南京凯基生物科技发展有限公司;GLUT2和β-actin抗体购自Abcam;TSC2、p-TSC2、4EBP1、p-4EBP1、S6和p-S6抗体购自CST;辣根过氧化物酶标记的IgG II 抗购自北京中杉金桥公司;BCA蛋白浓度检测试剂盒购自Thermo。
2主要方法
2.1细胞培养A549和NCI-H1650细胞培养于含10% FBS、1%青霉素和1%链霉素的高糖型DMEM培养基中,置于37 ℃、5%、CO2及完全湿度的培养箱中。待细胞融合度达到80%左右时进行细胞传代,按细胞密度传到相应细胞培养瓶中用于后续试验。
2.2MTT实验将生长良好的A549和NCI-H1650细胞经胰酶消化计数后,以4×104/well接种于96孔板中,细胞置于37 ℃、5% CO2孵箱中培养;24 h后弃掉培养基,分别加入200 μL不同终浓度的DHA DMEM完全培养基,同时设置对照组(含DSMO的DMEM完全培养基),置于37℃、5% CO2的培养箱中分别培养24 h和48 h。每孔加入20 μL 5 g/L MTT,继续培养4 h,弃上清,每孔加入150 μL DMSO,在摇床上轻微振荡5 min使结晶完全溶解,用酶标仪测定492 nm处各孔吸光值(A值)。重复3次,按下列公式计算细胞活力抑制率:细胞活力抑制率(%)=(对照组A均值-实验组A均值)/对照组A均值×100%。
2.3克隆形成实验将生长良好的A549和NCI-H1650细胞经胰酶消化后计数,细胞以200个/皿接种于10 cm细胞培养皿中,置于37 ℃、5% CO2培养箱中培养24 h;向培养皿中加入不同终浓度DHA(0、5、10、20、40 mg/L)DMEM培养基,设3个平行皿,置于37 ℃、5% CO2孵箱中培养15 d,直到通过出现肉眼能清晰观察到可见的细胞克隆;将培养液倒掉,用PBS洗涤,甲醇固定10 min,PBS洗涤,吉姆萨染色 10 min,自来水清洗,晾干后计数,按下列公式计算细胞克隆形成率:克隆形成率(%)=细胞克隆数平均值/铺板细胞总数×100%。
2.4流式细胞术实验生长良好的A549和NCI-H1650细胞经消化后接种到6孔板中,培养24 h后弃掉培养基,加入不同终浓度DHA(0、5、10、20、40 mg/L)DMEM培养基作用48 h,然后用PBS洗涤3次,无EDTA的胰酶消化,离心收集细胞,按照Annexin V/PI试剂盒说明书操作,先加入500 μL的binding buffer重悬细胞,再加入5 μL FITC标记的Annexin V和5 μL PI混匀,室温下避光孵育15 min,运用流式细胞仪测定细胞凋亡。
2.5葡萄糖和乳酸含量的测定生长良好的A549和NCI-H1650细胞经消化后以细胞密度为2.5×105/well接种到12孔板中,培养24 h后弃掉培养基,加入不同终浓度的DHA(0、10、20、40 mg/L)高糖型DMEM培养基分别作用24 h和48 h。收集培养液,12 000 r/min离心10 min去除细胞碎片,收集上清,用于葡糖糖和乳酸含量的测定。运用比色法按照氧化酶法-葡萄糖测定试剂盒说明书测定葡萄糖含量(nmol/L);乳酸含量测定前,用乳酸测定缓冲液将细胞上清稀释100倍,运用比色法按照乳酸测定试剂盒使用说明书测定乳酸含量(nmol/L)。
2.6ATP含量测定A549和NCI-H1650细胞处理条件同实验2.5,作用24 h,细胞经PBS洗涤2次,离心收集细胞。按照ATP生物发光检测试剂盒说明书,向细胞中加入ATP释放裂解液吹打混匀,冰上裂解30 min。再加入萤光素酶溶液,漩涡振荡1 min,取100 μL于反应管中,立即用荧光分光光度计测定荧光密度,通过标准曲线法计算ATP含量。
2.7细胞转染GST-GLUT2和GST-Rheb重组表达载体由本实验室构建。将生长良好的A549和NCI-H1650细胞以1×106/well接种到6孔板中,待细胞融合度达到80%左右时按照脂质体2000说明书进行转染。将5 μL脂质体2000和2 μg质粒溶解于无血清OptiMEM培养液中,轻轻混匀,室温静置10min后加入A549和NCI-H1650细胞中,培养4 h后换成完全培养基,未转染的细胞为空白对照,培养48 h用于后续实验。
2.8Western blotA549和NCI-H1650细胞经不同终浓度DHA作用不用时间,特定时间收集细胞。细胞沉淀中加入RIPA细胞裂解液(50 mmol/L Tris-HCl pH 7.5,150 mmol/L NaCl,1% NP-40,0.5% 脱氧胆酸钠,0.1% SDS),超声破碎、12 000 r/min,4℃离心10 min,按照BCA试剂盒说明书测定总蛋白浓度。每个样本以30 μg总蛋白进行SDS-PAGE,将蛋白转移到PVDF膜上,5%脱脂奶粉室温封闭1 h,分别孵育 I 抗(GLUT2、GLUT4、TSC2、p-TSC2、4EBP1、p-4EBP1、S6、p-S6和β-actin),4 ℃过夜。洗膜、孵育HRP-IgG II抗,室温1 h。ECL显影后扫描,蛋白相对表达量经内参照归一化后经Quantity-One软件分析。
2.9葡萄糖对细胞增殖和凋亡的影响A549和NCI-H1650细胞培养在无葡萄糖的DMEM培养基(含10% FBS、1%青霉素和1%链霉素),传代培养3 d。分别以4×104/well和4.5×105/well接种于96孔板和6孔板中,细胞贴壁后加入不同葡萄糖浓度(0、2、4、8 nmol/L)+40 mg/L DHA的DMEM培养基(不含葡萄糖,含10% FBS、1%青霉素和1%链霉素),继续作用48 h。按照MTT法,克隆形成实验和Annexin V/PI试剂盒说明书测定细胞活性、克隆形成和细胞凋亡。
3统计学处理
每个实验进行3次独立重复实验。应用SPSS 17.0统计软件进行相关数据分析,结果用均数±标准误(mean±SEM)表示,两组之间的比较采用t检验,多组间的比较采用单因素方差分析(one-way ANOVA)。以P<0.05为差异有统计学意义。
结果
1DHA抑制NSCLC细胞活力和克隆形成
将不同浓度的DHA处理A549和NCI-H1650细胞24 h和48 h后,运用MTT法和克隆形成实验测定细胞活力和细胞克隆形成能力。结果表明DHA能显著抑制A549和NCI-H1650的细胞活力(P<0.05),并具有时间和剂量依赖效应,DHA对A549细胞活力的半数抑制浓度分别为41.3 mg/L(24 h)和27.5 mg/L(48 h),DHA对NCI-H1650细胞活力的IC50分别为42.3 mg/L(24 h)和38.7 mg/L(48 h)。克隆形成实验进一步证实DHA能显著抑制A549和NCI-H1650细胞的克隆形成能力(P<0.05),随着DHA浓度的增加,细胞的克隆形成率逐渐降低,见图1。
2Annexin V-FITC染色和流式细胞术测定DHA诱导的NSCLC细胞凋亡
随着DHA浓度的增加,早期细胞凋亡率无显著变化,晚期细胞凋亡率与DHA浓度呈剂量依赖效应;DHA处理的A549和NCI-H1650细胞凋亡率显著高于对照组(P<0.01),见图2。
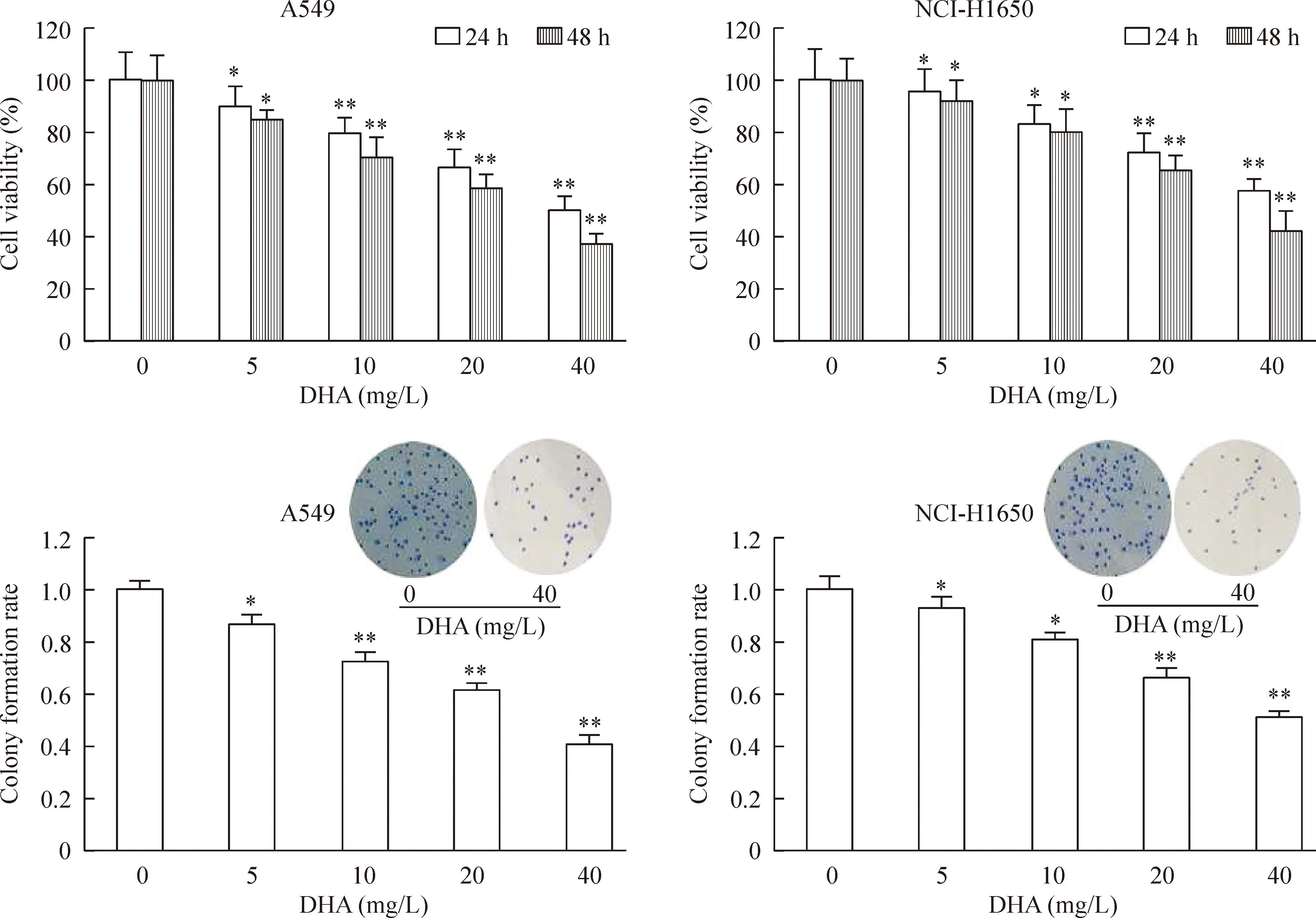
Figure 1.DHA inhibited cell growth and colony formation in A549 cells and NCI-H1650 cells. Mean±SEM.n=6.*P<0.05,**P<0.01vs0 mg/L group.
图1DHA抑制A549和NCI-H1650细胞活力和克隆形成能力
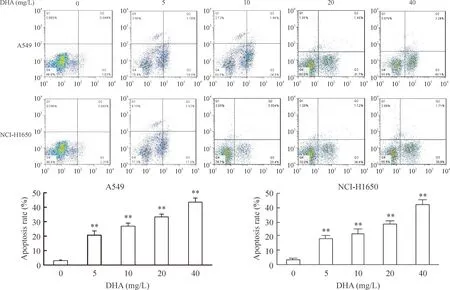
Figure 2.DHA induced apoptosis in A549 cells and NCI-H1650 cells determined by flow cytometry with Annexin V-FITC/PI staining. Mean±SEM.n=3.**P<0.01vs0 mg/L group.
图2DHA诱导A549和NCI-H1650细胞凋亡
3DHA抑制NSCLC细胞的糖酵解代谢
DHA能抑制A549和NCI-H1650细胞对葡萄糖的摄入,经DHA作用的NSCLC细胞其葡萄糖摄入量显著低于对照组(P<0.01),并呈时间和剂量依赖效应。10 mg/L DHA对A549和NCI-H1650细胞中ATP和乳酸含量无影响;当DHA浓度大于20 mg/L时,细胞内的ATP和乳酸含量显著降低(P<0.01),并呈剂量依赖效应。
Figure 3.DHA inhibited glycolytic metabolism in A549 cells and NCI-H1650 cells. Mean±SEM.n=3.*P<0.05,**P<0.01vs0 mg/L group.
图3DHA抑制A549和NCI-H1650细胞的糖酵解代谢
4DHA通过抑制PI3K通路活性和GLUT2表达抑制糖酵解代谢
将A549和NCI-H1650细胞经DHA作用48 h后,Western blot检测PI3K信号通路中关键蛋白(4EBP1和TSC-2)的表达。结果发现DHA能诱导4EBP1和TSC-2蛋白磷酸化,随着DHA浓度的增加,4EBP1和TSC-2蛋白磷酸化水平逐渐降低,呈现剂量依赖效应。然后用40 mg/L DHA作用A549细胞不同时间(0~24 h)后发现4EBP1和TSC-2蛋白磷酸化水平逐渐降低,并呈时间-依赖效应。同时,研究DHA对GLUT2 和 GLUT4表达的影响,结果显示DHA处理的NSCLC细胞中GLUT2的表达显著下调,并呈剂-量依赖效应,而GLUT4的表达无明显变化。本研究发现,与对照组比较,DHA显著诱导A549细胞凋亡(18 h和24 h)(P<0.05),而6 h和12 h的细胞凋亡率与对照组比较差异无统计学意义。A549细胞对葡萄糖摄入量显著低于对照组,并具有时间依赖效应,见图4。
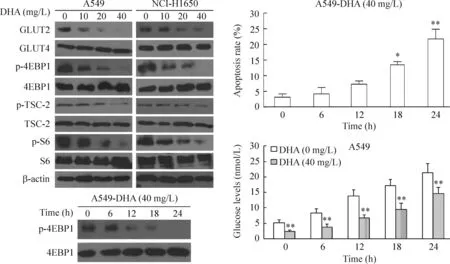
Figure 4.DHA suppressed glycolytic metabolism via inhibiting PI3K pathway activation and GLUT2 expression. Mean±SEM.n=3.*P<0.05,**P<0.01vs0 mg/L group.
图4DHA通过抑制PI3K通路活性和GLUT2表达抑制糖酵解代谢
5上调PI3K通路活性促进NSCLC细胞的糖酵解代谢和细胞增殖
上述研究显示DHA抑制糖酵解代谢和NSCLC细胞增殖与PI3K活性降低相关,而增强PI3K的活性能否减弱由DHA对糖酵解代谢和NSCLC细胞增殖的抑制作用?本研究发现A549细胞中Rheb的过表达能促进GLUT2的表达和4EBP1蛋白磷酸化;相反,DHA能抑制GLUT2的表达和4EBP1蛋白磷酸化,提示Rheb过表达能激活PI3K信号通路,而DHA能抑制Rheb对PI3K通路的激活。同时,DHA显著抑制A549细胞增殖和细胞对葡萄糖的摄入以及ATP的产生,而Rheb的过表达能抑制DHA的这一功能,见图5。
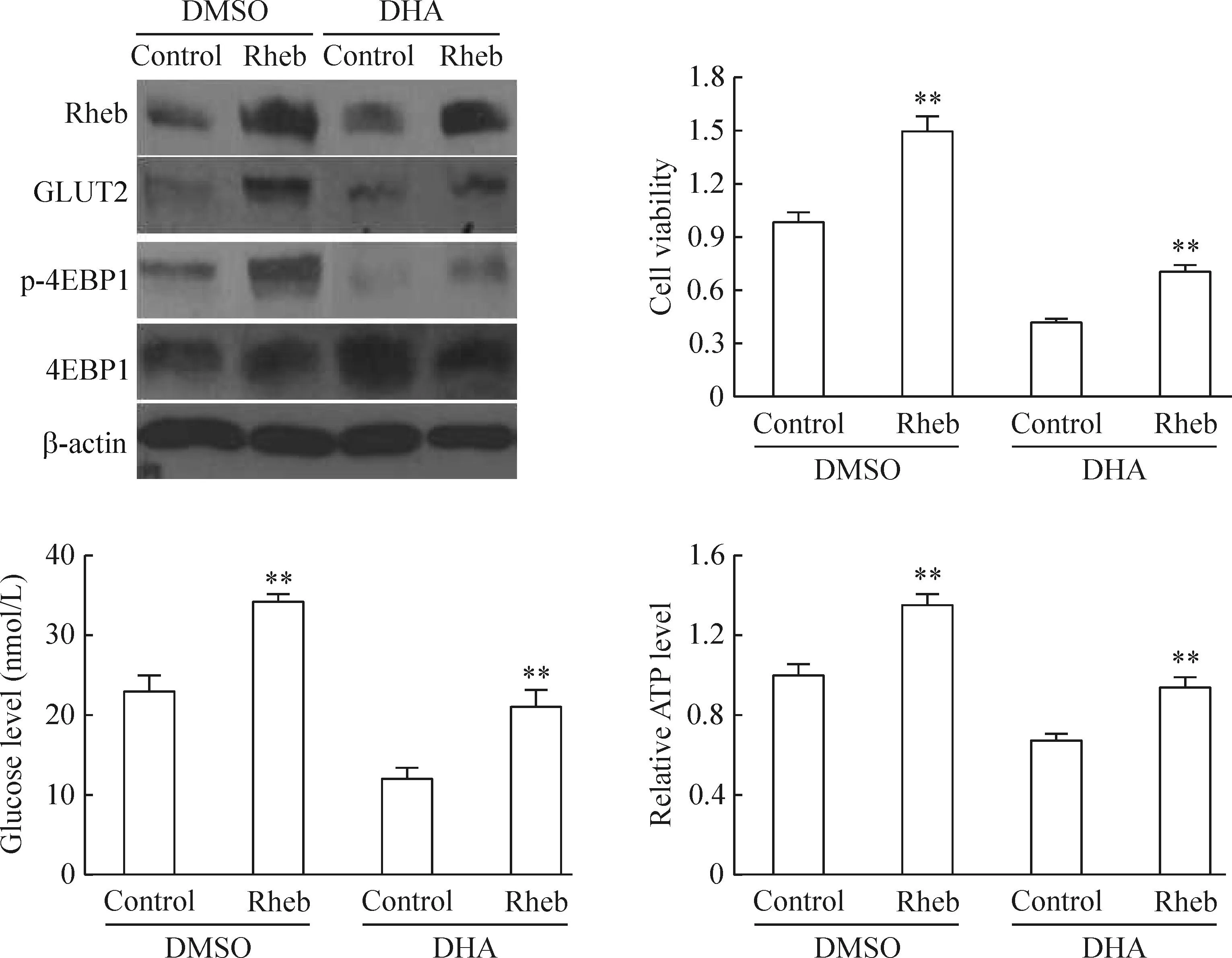
Figure 5.Upregulated PI3K pathway activation promoted glycolytic metabolism and growth in A549 cells. A549 cells were transfected with control or Rheb vector, and then treated with the indicated concentration of DHA for 48 h. Mean ± SEM.n=3.**P<0.01vscontrol group.
图5上调PI3K通路活性促进A549细胞糖酵解代谢和提高细胞活力
6GLUT2过表达抑制由DHA介导的NSCLC细胞毒性
以上的研究表明DHA通过下调GLUT2的表达抑制NSCLC细胞对葡萄糖的摄入,从而诱导细胞毒性。基于这一发现,有必要研究GLUT2的过表达能否抑制DHA对NSCLC细胞的毒性作用。结果显示外源性GST-GLUT2质粒转染的A549和NCI-H1650细胞中GLUT2的表达显著高于对照组(未转染细胞)。转染的NSCLC细胞培养48 h后,高表达GLUT2的细胞存活率显著高于对照组(未转染细胞)(P<0.01);当细胞经40 mg/L DHA作用48 h后,高表达GLUT2的细胞存活率显著低于溶剂对照组(DMSO处理的细胞)(P<0.01),但仍显著高于对照组(DHA未处理的细胞),见图6。
7高浓度葡萄糖能抑制DHA对NSCLC细胞的毒性作用
本研究提示DHA通过抑制NSCLC细胞对葡萄糖的摄入和糖酵解代谢从而诱导细胞毒性作用。为了进一步证实这一结果,用不同浓度的葡萄糖培养A549和NCI-H1650细胞,观察葡萄糖缺乏能否增强DHA的细胞毒性作用。结果如图7所示,当培养基中无葡萄糖存在时(0 mmol/L),细胞存活率为59.4%(A549细胞)和62.5%(NCI-H1650细胞),显著高于DHA(40 mg/L)作用下细胞存活率(P<0.01);随着葡萄糖浓度的增加(2、4、8 mmol/L),细胞存活率逐渐升高。当葡萄糖浓度为8 mmol/L时,细胞存活率几乎不受影响;克隆形成实验也能观察到相似的结果,提示葡萄糖匮乏能诱导NSCLC细胞毒性,而葡萄糖能减弱DHA对细胞的毒性作用。高浓度葡萄糖能否抑制由DHA诱导NSCLC细胞凋亡,A549和NCI-H1650细胞经不同浓度葡萄糖培养48 h后,Annexin V-FITC/PI染色结果显示,无葡萄糖存在时(0 mmol/L)细胞凋亡率为25%(A549细胞)和20%(NCI-H1650细胞),而40 mg/L DHA单独作用下细胞凋亡率为52%(A549细胞)和61%(NCI-H1650细胞),随着葡萄糖浓度的增加细胞凋亡率逐渐降低。
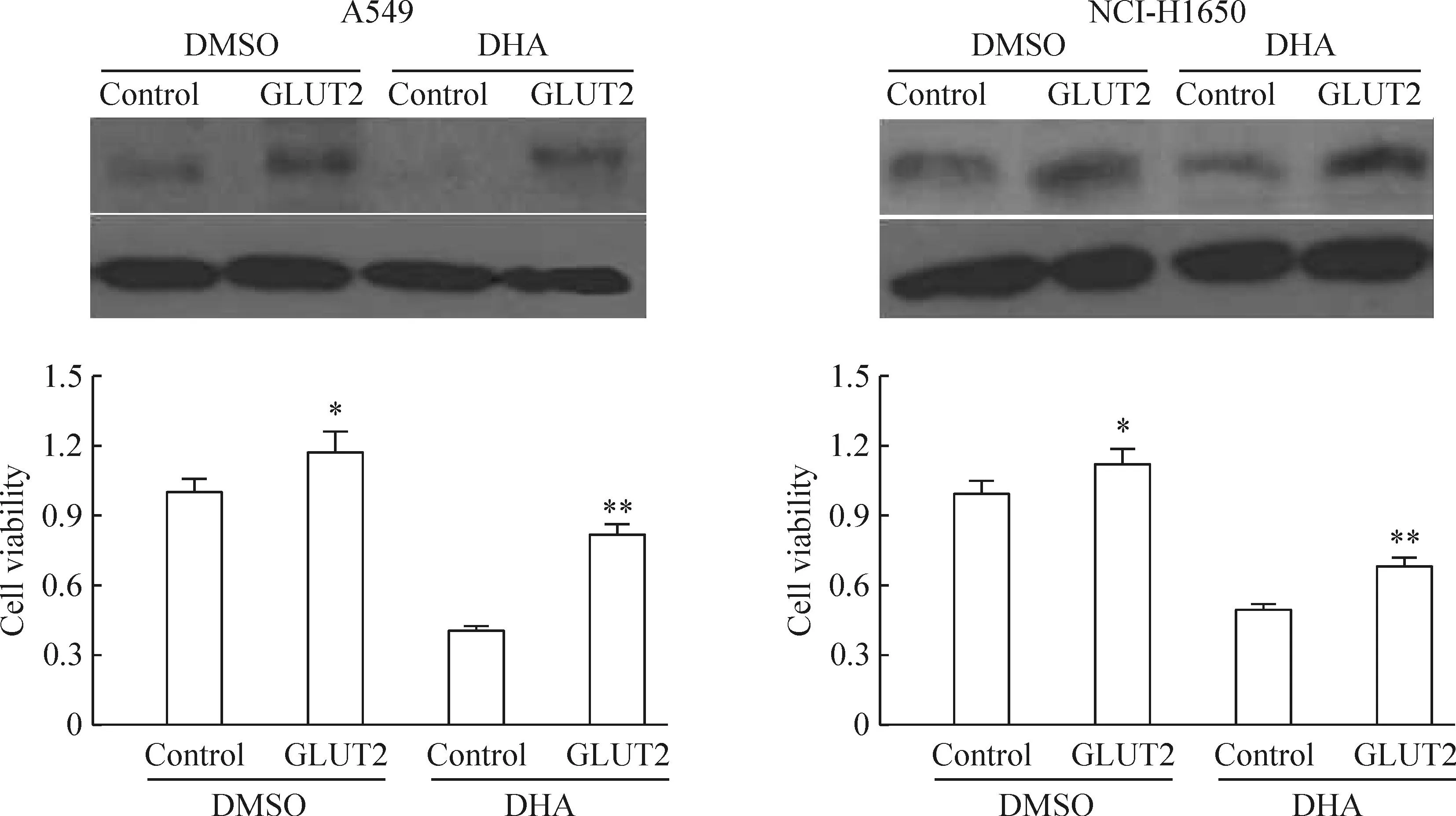
Figure 6.Overexpression of GLUT2 inhibited cell death triggered by DHA in A549 cells and NCI-H1650 cells. A549 cells and NCI-H1650 cells were transfected with control or GLUT2 vector, and then treated with the indicated concentration of DHA for 48 h. Mean±SEM.n=3.*P<0.05,**P<0.01vscontrol group.
图6GLUT2过表达抑制由DHA介导的NSCLC细胞死亡
讨论
最新的研究发现DHA能抑制肝癌、胰腺癌和前列腺癌细胞增殖并诱导细胞凋亡[3-4,16]。本研究结果中,DHA具有抑制A549和NCI-H1650细胞活力和克隆形成能力以及诱导细胞凋亡的活性,且呈剂-量依赖效应,与上述文献报道一致。此外,本研究首次发现DHA能够抑制NSCLC细胞的糖酵解代谢。有研究发现PI3K通路在细胞糖代谢过程中发挥着重要的调控作用,激活mTOR通路能够促进肿瘤细胞对葡萄糖的摄入从而促进葡萄糖代谢[11-12]。在卵巢癌细胞和横纹肌肉瘤细胞中的研究也发现DHA能抑制mTOR通路活性[17-18]。本研究在NSCLC细胞中的研究发现DHA能抑制PI3K通路活性和葡萄糖代谢以及A549细胞对葡萄糖的摄入,并呈时间-依赖效应。此外,40 mg/L DHA作用12 h的A549细胞的凋亡率与对照组比较差异无统计学意义,提示DHA抑制糖酵解代谢是通过抑制PI3K通路活性,而不是激活细胞凋亡通路。
有研究表明GLUT在大多数肿瘤细胞中高表达,而在正常上皮细胞中不表达[13-14]。促进GLUT的表达能够加速细胞对葡萄糖摄取从而满足肿瘤细胞对葡萄糖的需求[10]。早期的研究发现雷帕霉素抑制mTOR通路活性进而抑制GLUT1的表达最终抑制细胞对葡萄糖的摄入[15]。我们在NSCLC细胞中的研究发现DHA能抑制GLUT2的表达,而GLUT2的高表达能够减轻DHA对NSCLC细胞的抑制作用。在血管平滑肌细胞中的研究发现GSK-3/TSC2/mTOR通路能够调控GLUT1的表达[19]。糖酵解代谢受多种蛋白激酶调节,有研究表明PI3K对糖酵解代谢起正调控作用[11]。为揭示DHA是否通过抑制PI3K通路活性进而抑制糖酵解代谢,最终发挥细胞毒性作用,将A549和NCI-H1650细胞经DHA作用48 h后检测PI3K信号通路中关键蛋白(4EBP1和TSC-2)的表达情况。Western blot结果显示NSCLC细胞中PI3K活性下调(p-4EBP1和p-TSC2表达降低),同时发现GLUT2的表达被抑制且具有PI3K活性依赖关系,而上调PI3K的活性(A549细胞中Rheb的过表达)能促进GLUT2的表达和4EBP1蛋白磷酸化,提示PI3K通路调控GLUT2的表达。因此,本研究表明DHA通过抑制PI3K通路活性从而下调GLUT2的表达,最终抑制细胞对葡萄糖的摄入。
癌细胞内的生物合成速度和葡萄糖代谢反应加快,使得癌细胞经常遭遇代谢应激和营养匮乏[9]。因此,本研究将NSCLC细胞培养在低浓度葡萄糖或无葡萄糖的培养基中,观察NSCLC细胞遭遇代谢应激时的生理反应。结果表明,DHA作用下葡萄糖缺乏能诱导NSCLC细胞毒性;DHA和葡萄糖匮乏联合作用的NSCLC细胞死亡率显著高于DHA单独作用下的细胞死亡率。癌细胞在葡萄糖匮乏或葡萄糖浓度较低时ATP含量显著降低,线粒体死亡通路被激活从而促进氧化应激和激活MAPK信号通路[20-22]。本研究结果显示,GLUT2高表达抑制DHA对A549和NCI-H1650细胞的毒性作用,而高浓度葡萄糖能抑制DHA对NSCLC细胞的毒性作用;研究证实GLUT2的过表达增加葡萄糖的转运速度,促进细胞对葡萄糖的摄取[10]。本研究发现DHA作用下NSCLC细胞中ATP和乳酸含量显著降低,并呈浓度依赖效应,提示DHA抑制NSCLC细胞对葡萄糖的摄入是其诱导细胞毒性作用可能的分子机制。
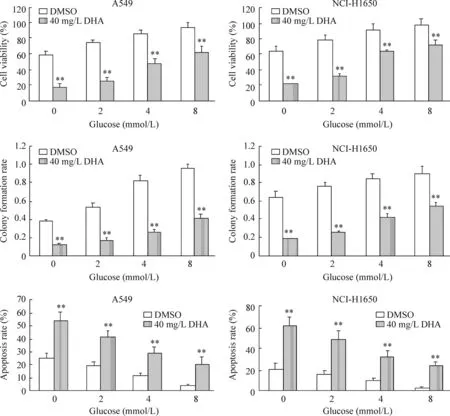
Figure 7.High glucose restrained DHA-induced cytotoxicity in A549 cells and NCI-H1650 cells. Mean±SEM.n=3.**P<0.01vsDMSO-treated cells.
图7高浓度葡萄糖抑制由DHA诱导的NSCLC细胞毒性作用
DHA能诱导NSCLC细胞凋亡,并具有时间和剂量依赖效应,但DHA诱导NSCLC细胞凋亡的分子机制仍不清楚。研究表明DHA通过线粒体依赖性通路诱导结直肠癌细胞凋亡[23]。最新的研究发现DHA与电离辐射联合作用显著诱导A549细胞凋亡,且不依赖于线粒体依赖性通路[24]。DHA与阿霉素联合作用通过诱导细胞凋亡从而对乳腺癌细胞产生细胞毒性作用[25]。本研究也证实,DHA诱导NSCLC细胞凋亡从而发挥细胞毒性作用,而葡萄糖能减弱DHA对NSCLC细胞毒性和DHA介导的NSCLC细胞凋亡。然而,对DHA诱导NSCLC细胞凋亡的具体分子机制有必要做深入研究。
综上所述,本研究表明DHA显著抑制NSCLC细胞系A549和NCI-H1650细胞的增殖和克隆形成能力以及诱导细胞凋亡,并具有时间-剂量依赖效应。此外,本研究首次发现DHA能抑制NSCLC细胞的糖酵解代谢,且DHA对糖酵解代谢的抑制作用与PI3K通路活性和GLUT2表达下调相关,这一发现可能是DHA诱导NSCLC细胞毒性作用的分子机制,提示DHA有望成为治疗NSCLC潜在的新型抗肿瘤药物。然而,DHA抗肿瘤功能的生物学信息有待进一步研究。
[参考文献]
[1]石歆. 桔皮素对人非小细胞肺癌细胞生长及侵袭的影响[J]. 中国病理生理杂志, 2015, 31(3): 452-456.
[2]叶燕霞, 曾耀英, 黄秀艳, 等. 双氢青蒿素对小鼠 T 细胞的免疫抑制作用[J]. 中国病理生理杂志, 2010, 26(3): 417-423.
[3]Zhang S, Ma Y, Jiang J, et al. Inhibition of urokinase-type plasminogen activator expression by dihydroartemisinin in breast cancer cells[J]. Oncol Lett, 2014, 7(5):1375-1380.
[4]Kong R, Jia G, Cheng ZX, et al. Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in pancreatic cancer cells via ROS-mediated up-regulation of death receptor 5[J]. PLoS One, 2012, 7(5):e37222.
[5]Zhou HJ, Zhang JL, Li A, et al. Dihydroartemisinin improves the efficiency of chemotherapeutics in lung carcinomasinvivoand inhibits murine Lewis lung carcinoma cell line growthinvitro[J].Cancer Chemother Pharm, 2010, 66(1):21-29.
[6]周喜汉, 黄赞松, 向发良. 中药抗结直肠癌作用的研究进展[J]. 世界华人消化杂志, 2013, 21(18): 1720-1725.
[7]Wang SJ, Sun B, Cheng ZX, et al. Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-κB pathway[J]. Cancer Chemother Pharm, 2011, 68(6):1421-1430.
[8]Zhao YG, Wang Y, Guo Z, et al. Dihydroartemisinin ameliorates inflammatory disease by its reciprocal effects on Th and regulatory T cell function via modulating the mammalian target of rapamycin pathway[J]. J Immunol, 2012, 189(9):4417-4425.
[9]Ariffin AB, Forde PF, Jahangeer S, et al. Releasing pressure in tumors: what do we know so far and where do we go from here? A review[J]. Cancer Res, 2014, 74(10):2655-2662.
[10]Li X, Luo H, Paul SC, et al. Downregulation of the expression of GLUT1 plays a role in apoptosis induced by sodium butyrate in HT-29 cell line[J]. Int J Mol Sci, 2006, 7(3):59-70.
[11]Laplante M, Sabatini DM. mTOR signaling in growth control and disease[J]. Cell, 2012, 149(2):274-293.
[12]Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer[J]. Curr Opin Genet Dev, 2013, 23(3):53-62.
[13]王玮, 蔡建辉, 马毓梅, 等. GLUT4 在多囊卵巢综合征大鼠子宫内膜中的表达及意义[J]. 中国病理生理杂志, 2007, 23(3): 587-590.
[14]Cai Y, Zhai JJ, Feng BB, et al. Expression of glucose transporter protein 1 and p63 in serous ovarian tumor[J]. J Obstet Gynaecol Re, 2014, 40(7):1925-1930.
[15]Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity[J]. Science, 2014, 345(6204):1250684.
[16]Buller CL, Loberg RD, Fan MH, et al. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression[J]. Am J Physiol Cell Physiol, 2008, 295(3):C836-C843.
[17]He Q, Shi J, Shen XL, et al. Dihydroartemisinin upregulates death receptor 5 expression and cooperates with TRAIL to induce apoptosis in human prostate cancer cells[J]. Cancer Biol Ther, 2010, 9(10):819-824.
[18]Feng X, Li L, Jiang H, et al. Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: involvement of apoptosis and autophagy[J]. Biochem Biophys Res Commun, 2014, 444(3):376-381.
[19]Odaka Y, Xu B, Luo Y, et al. Dihydroartemisinin inhibits the mammalian target of rapamycin-mediated signaling pathways in tumor cells[J]. Carcinogenesis, 2014, 35(1):192-200.
[21]El Mjiyad N, Caro-Maldonado A, Ramirez-Peinado S, et al. Sugar-free approaches to cancer cell killing[J]. Oncogene, 2011, 30(3): 253-264.
[22]Raina K, Agarwal C, Wadhwa R, et al. Energy deprivation by silibinin in colorectalcancer cells: a double-edged sword targeting both apoptotic and autophagic machineries[J]. Autophagy, 2013, 9(5):697-713.
[23]Lu M, Sun L, Zhou J, et al. Dihydroartemisinin induces apoptosis in colorectal cancer cells through the mitochondria-dependent pathway[J]. Tumour Biol, 2014, 35(6):5307-5314.
[24]Chen T, Chen M, Chen J. Ionizing radiation potentiates dihydroartemisinin-induced apoptosis of A549 cells via a caspase-8-dependent pathway[J]. PLoS One, 2013, 8(3): e59827.
[25]Wu GS, Lu JJ, Guo JJ, et al. Synergistic anti-cancer activity of the combination of dihydroartemisinin and doxorubicin in breast cancer cells[J]. Pharmacol Rep, 2013, 65(2):453-459.
(责任编辑: 陈妙玲, 罗森)

