纤维性肾小球肾炎
2012-11-26曾彩虹朱小东陈惠萍刘志红
曾彩虹 黄 倩 朱小东 陈惠萍 刘志红
病例摘要
病史21岁男性,因“反复水肿、尿检异常2年”于2011-10-17入院。
患者于2009年6月上感后出现颜面部及双下肢水肿,当地医院查尿蛋白3+、隐血2+,伴低白蛋白血症(具体不详),肾功能正常,无贫血,血压波动于130~140/80~90 mmHg,予泼尼松 50mg/d,逐渐减量,共服半年,其间加服中药偏方,监测尿蛋白最低降至±,但时有反复;此后长期服用中药汤剂治疗。2010年11月曾行环磷酰胺(CTX)冲击治疗(1次/周×3次,剂量不详),复查尿蛋白波动于2+~3+。2011年7月劳累后水肿加重,查尿蛋白定量6.09 g/24h,隐血 +,血清白蛋白(Alb)15.7 g/L,肾功能正常,胆固醇7.64 mmol/L,三酰甘油2.41 mmol/L,ENA多肽谱中抗SSB弱阳性,余阴性;乙肝标志物阴性。当地医院行肾活检诊断为“特殊蛋白沉积肾病”。同年8月2日再次服泼尼松55mg/d,同时以硝苯地平缓释片及缬沙坦降压治疗,水肿较前消退,尿蛋白定量降至1.4 g/24h,但Alb无升高。10月14日患者于我科就诊,并收治入院。病程中无皮疹、关节痛及脱发,夜尿0~1次/晚。精神、饮食、睡眠尚可,体重增加2 kg。
既往史、家族史:平素易感冒,余无特殊。父母体健,否认家族中同样疾病患者。
体格检查 体温 37℃,脉搏80次/min,呼吸20次/min,血压 133/79 mmHg,体质量指数 20.3 kg/m2。神清、精神好,发育正常,全身皮肤、巩膜无黄染,浅表淋巴结未及肿大,双肺呼吸音清,未闻及干、湿性啰音,心脏听诊未及异常,腹平软,全腹无压痛及反跳痛,肝、脾肋下未及,肝、肾区无叩痛,双下肢轻度水肿。
实验室检查
血常规 血红蛋白122 g/L,白细胞12.2×109/L,中性粒细胞/淋巴细胞81%/15%,血小板286×109/L。
尿液 尿蛋白定量0.46~1.34 g/24h,尿沉渣红细胞计数1.0万/ml;C3 2.4mg/L,α2巨球蛋白(α2-MG)2.11mg/L;N-乙酰-β-D-氨基葡萄糖苷酶(NAG)35.53 U/(g·Cr),视黄醇结合蛋白(RBP)<0.5mg/L,溶菌酶(Lyso)<0.5mg/L,中性粒细胞明胶酶相关脂质运载蛋白(NAGL)10.85μg/L,肾损伤分子1(KIM-1)0.08μg/L,白细胞介素18(IL-18)17.11 ng/L;尿渗量 627 mOsm/(Kg·H2O),尿氨基酸150.6mg/24h;尿 κ轻链 57.58mg/L,λ轻链11.09mg/L,本周蛋白阴性。
血生化 Alb 20.0 g/L,球蛋白15.6 g/L,尿素氮 8.50 mmol/L,肌酐 57.5 μmd/L,尿酸 511 μmol/L,胱抑素C 1.79mg/L,谷丙转氨酶23 U/L,谷草转氨酶17 U/L,总胆固醇8.48 mmol/L,三酰甘油 1.35 mmol/L,钾3.95 mmol/L,钠140.8 mmol/L,氯108.1 mmol/L,二氧化碳总量26.5 mmol/L,钙1.85 mmol/l,磷 1.25 mmol/L,C 反应蛋白 1.0mg/L。血κ轻链16.62mg/L,λ轻链16.57mg/L,血清免疫固定电泳阴性。糖化血红蛋白5.5%。
免疫球蛋白及自身抗体 ANA、A-dsDNA阴性。ENA多肽谱中SSB+,余阴性;抗心磷脂抗体阴性。补体C3 0.904 g/L,C4 0.171 g/L。冷球蛋白74.9mg/L。IgG 3.14 g/L,IgA 1.4 g/L,IgM 1.18 g/L,链球菌溶血素“O”26.5 IU/ml,类风湿因子 <20 IU/ml。乙型肝炎(HRV)、丙型肝炎(HCV)、梅毒及艾滋抗体检测均阴性。
其他 外周血淋巴细胞亚群 CD4 421个/μl,CD8 596 个/μl,CD3 1 045 个/μl,CD20 255 个/μl,Treg 22 个/μl。
辅助检查
双肾B超 左肾:103 mm×50 mm×51 mm;右肾111 mm×49 mm×55 mm,双肾轮廓规则,包膜连续完整;双肾内未见肾盂肾盏扩张。
肝、胆、胰、脾B超 肝周及下腹部可探及大量液性暗区。
胸片 (1)左肺下野炎症可能;(2)左侧少量胸腔积液。
其他 心电图正常。甲状腺超声未见明显异常。
肾活检病理
光镜 16个肾小球,正切肾小球体积增大,增宽的系膜区及外周袢被大量PAS弱阳性(图1A)、嗜亮绿的无定形物质占据,系膜细胞、内皮细胞增生不明显;毛细血管袢开放欠佳,数处见透明滴,囊壁增厚、节段分层,囊周纤维化。PASM-Masson:系膜区及外周袢嗜银性减弱,呈嗜亮绿色(图1B)。肾小管间质慢性轻度病变,灶性小管萎缩、基膜增厚,管腔内较多蛋白管型,少量非萎缩肾小管上皮细胞细空泡变性,间质灶性纤维化,见单个核细胞浸润。动脉未见明确病变。肾组织刚果红染色阴性。
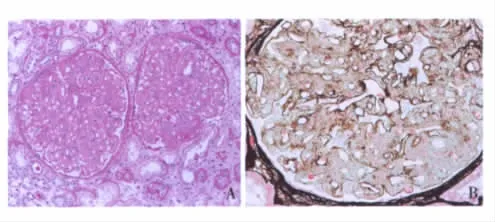
图1 A:增宽的肾小球系膜区和外周袢被PAS淡染的物质代替(PAS,×200);B:肾小球系膜区及外周袢嗜银性减弱,呈嗜亮绿色(PASM-Masson,×400)
免疫荧光 IgG++、IgM+、C3++、C1q+,呈颗粒状弥漫分布于系膜区及血管袢(图2)。IgA+少量呈细颗粒状沉积于血管袢。C3球囊壁阳性,纤维蛋白阴性。肾组织IgG亚型染色示IgG1++、IgG2++、IgG4++,呈颗粒状弥漫分布于系膜区及血管袢。IgG3阴性。肾组织轻链染色示κ轻链++、λ轻链+,呈颗粒状弥漫分布于系膜区及血管袢(图3)。纤维连接蛋白染色阴性;胶原Ⅲ染色肾小球系膜区节段少量阳性。肾组织Ⅳ型胶原 α3、α5链染色肾小球基膜(GBM)、肾小管基膜(TBM)均正常。
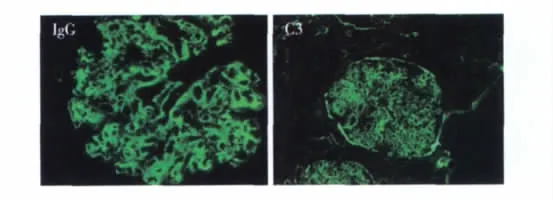
图2 免疫荧光染色IgG++、C3++,呈颗粒状弥漫分布于肾小球系膜区及血管袢(IF,×400)
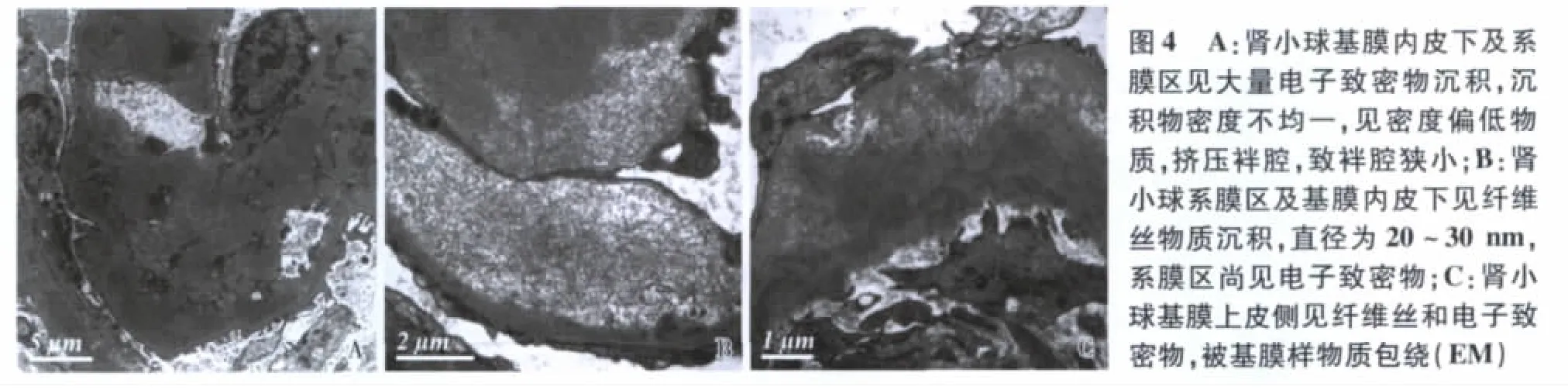
电镜 观察1个肾小球。肾小球系膜区增宽,基膜样物质增多,系膜区、系膜旁区及节段内皮下见大量电子致密物,沉积物密度不均一,见密度偏低物质挤压袢腔,致袢腔狭小(图4A),有的沉积物内见细小脂性空泡及淡染物质。GBM节段分层,分层的GBM内及内皮下亦见密度减低的物质,高倍镜下见排列紊乱的细纤维丝,直径20~30 nm(图4B)有的沉积物分布于GBM上皮侧,周围见GBM样物质分隔或包绕。高倍镜下上皮侧的致密物内见类似细纤维丝(图4C)。数处上皮侧致密物密度减低,略显吸收。少数袢节段扭曲、塌陷,并连接成片。类似纤维丝也见于系膜区。肾小球足细胞足突广泛融合,少量微绒毛化。
小结:符合纤维性肾小球肾炎。

讨 论
该患者系青年男性,临床以大量蛋白尿,低白蛋白血症,少量镜下血尿为主要表现,肾功能正常,无关节痛、皮疹等肾外系统受累。ANA、ds-DNA、ANCA、HBV、HCV标记物均阴性,仅 SSB+,患者否认口干、眼干等症状。患者特征性的组织形态学改变包括肾小球内不嗜银的异常物质沉积,免疫荧光肾小球IgG、C3阳性,少量 IgM 和 C1q沉积,散在IgA阳性;电镜下观察到肾小球系膜区、GBM内皮下和上皮侧纤维丝状物质沉积伴电子致密物分布,纤维丝直径为20~30 nm。
纵观该患者的肾活检病理特点,最具特征性的是超微结构改变可见肾小球内纤维丝状物质沉积,因此诊断和鉴别诊断分析首先应从超微结构特点即纤维丝状物质沉积入手。纤维丝物质沉积最常见于肾淀粉样变性,故这类患者在光镜、免疫病理及电镜检查的基础上,还应行刚果红染色,如刚果红染色阳性提示肾淀粉样变性,可进一步行淀粉样物质的相关染色,以对淀粉样变性进行分型。如患者刚果红染色阴性,排除肾淀粉样变性。对于刚果红染色阴性的患者,再根据免疫球蛋白阳性和阴性进行鉴别诊断。纤维丝或管状物质沉积伴免疫球蛋白IgG阳性的疾病包括纤维性肾小球肾炎、免疫管状肾小球病、冷球蛋白血症和部分增生性肾小球肾炎伴单克隆IgG沉积。冷球蛋白血症肾损害常为膜增生性病变,半数患者袢腔内见PAS阳性的冷球蛋白栓塞,超微结构观察为厚壁的环状、弯曲的微管状物,直径20~35 nm,横切面呈中空管状,其分布成对,随机或成簇。该患者血冷球蛋白水平正常,沉积物也非微管状物,而是纤维丝状物,故排除冷球蛋白血症肾损害。部分增生性肾小球肾炎伴单克隆IgG沉积者也可出现微管状或晶格状物质[1],但沉积的免疫球蛋白为单一IgG亚型和单一轻链,而该患者沉积物为纤维丝状结构,且IgG亚型包括IgG1、IgG2、IgG4,κ轻链、λ轻链均阳性,故不支持增生性肾小球肾炎伴晶格状结构的单克隆IgG沉积的诊断。免疫管状肾小球病超微结构下沉积物呈中空微管状物,多数直径为30~60 nm,少数小至20 nm,大至90 nm,微管状物呈平行有序排列,有的排列较紧密,有的较松散,而该患者并未见中空微管状物,以纤维丝状物沉积为主(纤维丝直径为20~30 nm)伴电子致密物沉积,同时伴有IgG和轻链沉积,诊断考虑纤维性肾小球肾炎。
纤维性肾小球肾炎病理形态学变化多样,可有膜增生性病变、弥漫增生性病变、系膜增生,甚至膜性病变,但共同特点是可见无细胞结构的无定形物质分布于增宽的肾小球系膜区和肾小球外周袢,致肾小球毛细血管壁增厚。这些物质具有HE略淡染,PAS阳性的特性,银染色有助诊断。嗜银的胶原性基质与不嗜银的沉积物混杂导致增宽的系膜区、增厚的毛细血管壁可见“虫蚀样”改变。个别病例报道纤维性肾小球肾炎伴有新月体形成[2]。
免疫荧光染色在纤维性肾小球肾炎中具有鉴别诊断意义,因为纤维性肾小球肾炎常见免疫球蛋白和补体沉积于系膜区和(或)血管袢,单纯系膜区沉积时可表现为逗点状,外周袢沉积为类线样、假线样或飘带样[3]。沉积物最常见为IgG、C3,κ和λ轻链阳性。据文献报道纤维性肾小球肾炎仅少数(<10%)为单克隆免疫球蛋白沉积(一般为IgG κ),IgG 以 IgG4 和 IgG1 亚型最常见[4,5]。而该患者非单克隆性 IgG沉积,肾小球 IgG1、IgG2和IgG4均阳性,且κ和λ轻链同时阳性,因此IgG亚型染色只起到明确沉积物是否单克隆性质的作用,对诊断及鉴别诊断意义较小。
纤维性肾小球肾炎电镜下观察肾小球系膜区增宽,见无分枝,无序排列的纤维丝,直径10~30 nm,多数20 nm左右。纤维丝还分布于GBM内、内皮下和上皮侧。纤维丝分布区域常混有颗粒状或块状电子致密物,可能是由常规免疫致密物与纤维丝同时沉积所致,以致成为本病的一大特点。少数病例见纤维丝分布于TBM和管周毛细血管壁上,也有心脏和肺组织沉积的报道。
纤维性肾小球肾炎因其超微结构特点,需与多种疾病进行鉴别。免疫管状肾小球病沉积物的直径较大,多数>30 nm,且呈中空管状,平行排列。而淀粉样变性因其刚果红染色阳性较易鉴别。纤维丝沉积于上皮侧时应与膜性肾病鉴别,后者沉积物为颗粒状[6]。
该患者肾小球内有少量节段分布的胶原Ⅲ,还需与胶原纤维肾小球病和指甲-髌骨综合征进行鉴别,胶原纤维肾小球病特点为纤维见明暗交替带,纤维直径45~65 nm,广泛分布于肾小球系膜区和GBM内皮下,免疫荧光胶原Ⅲ染色肾小球弥漫阳性。指甲-髌骨综合征为GBM呈现虫蚀样病变,病变处见胶原纤维,伴明显的明暗交替带,胶原Ⅲ染色阳性,同时伴相应临床表现,而该患者胶原Ⅲ少量节段分布,且电镜下未见明暗交替的胶原Ⅲ,考虑胶原Ⅲ为非特异性的沉积。
纤维性肾小球肾炎是一种少见疾病,约占自体肾活检的0.5%~1%[4,7]。多累及中老年患者,男女发病率相似,主要表现为蛋白尿,半数以上达肾病范围蛋白尿。血尿(52%~60%)和高血压(71%~77%)较常见,肾功能不全的发生率高达 66%~72%[4,8]。血清学检查少数伴补体C3下降,抗核抗体低滴度阳性,部分患者存在 HCV感染[9-11]。而该患者仅21岁,临床也以蛋白尿为主,目前肾功能正常。故仅凭临床很难推测诊断。
纤维性肾小球肾炎的发病机制仍不清楚。免疫电镜证实纤维丝IgG、C3和P物质共沉积,而胶原Ⅳ、硫酸肝素糖蛋白和纤维连接蛋白阴性。提示纤维丝为免疫球蛋白来源。少数患者伴HCV感染,提示HCV导致的免疫复合物可形成纤维丝样结构[9,10]。因为从组织中尚未分离出这种纤维,故其具体结构仍未可知。也有报道纤维性肾小球肾炎伴发于骨髓瘤和意义未明的单克隆病(monoclonal gammopathy of undetermined significance,MGUS)[12,13]。
纤维性肾小球肾炎预后较差,肾活检后约半数在2~4年进展为终末期肾病,疾病进程与组织学类型相关,系膜增生者进展慢于膜增生性肾小球肾炎者。患者肾活检时血清肌酐水平高和间质纤维化程度重者预后差。目前尚无有效方法治疗纤维性肾小球肾炎,有文献报道利妥昔单抗能降低患者的蛋白尿14],肾移植后的复发率高达36%~47%[8,15]。
小结:纤维性肾小球肾炎为少见疾病,临床以蛋白尿为主要表现,部分伴肾功能不全。光镜下肾小球内无定形的不嗜银性物质沉积,免疫病理伴IgG和C3沉积,超微结构见纤维丝状物质和电子致密物沉积于肾小球系膜区、GBM内皮下、GBM内和上皮侧,多数患者预后较差。诊断的关键虽依赖于超微结构的观察,尚需综合光镜和免疫病理特点。
1 曾彩虹,朱小东.增生性肾小球肾炎伴晶格状结构的单克隆IgG沉积.肾脏病与透析肾移植杂志,2011,20(4):384-390.
2 Sharma P,Kuperman M,Racusen L,et al.Fibrillary glomerulonephritis presenting as rapidly progressive glomerulonephritis.Am JKidney Dis,2012,60(1):157 -159.
3 Sethi S,Adeyi OA,Rennke HG.A case of fibrillary glomerulonephritis with linear immunoglobulin G staining of the glomerular capillary walls.Arch Pathol Lab Med,2001,125(4):534 -536.
4 Rosenstock JL,Markowitz GS,Valeri AM,et al.Fibrillary and immunotactoid glomerulonephritis:Distinct entities with different clinical and pathologic features.Kidney Int,2003,63(4):1450 -1461.
5 Grove P,Neale PH,Peck M,et al.Monoclonal immunoglobulin G1-kappa fibrillary glomerulonephritis. Mod Pathol, 1998, 11(1):103-109.
6 Müller-Höcker J,Weiss M,Sitter T,et al.Fibrillary glomerulonephritis mimicking membranous nephropathy—a diagnostic pitfall.Pathol Res Pract,2009,205(4):265 -271.
7 Alpers CE, Kowalewska J. Fibrillary glomerulonephritis and immunotactoid glomerulopathy.J Am Soc Nephrol,2008,19(1):34-37.
8 Nasr SH,Valeri AM,Cornell LD,et al.Fibrillary glomerulonephritis:a report of 66 cases from a single institution.Clin J Am Soc Nephrol,2011,6(4):775 -784.
9 Ray S,Rouse K,Appis A,et al.Fibrillary glomerulonephritis with hepatitis C viral infection and hypocomplementemia.Ren Fail,2008,30(7):759-762.
10 Markowitz GS,Cheng JT,Colvin RB,et al.Hepatitis C viral infection is associated with fibrillary glomerulonephritis and immunotactoid glomerulopathy.JAm Soc Nephrol,1998,9(12):2244 -2252.
11 Fogo A,Qureshi N,Horn RG.Morphologic and clinical features of fibrillary glomerulonephritis versus immunotactoid glomerulopathy.Am JKidney Dis,1993,22(3):367 -377.
12 Park JH,Kim BR,Chun BG,et al.Coexistence of fibrillary glomerulonephritis in a patient with multiple myeloma.Intern Med,2012,51(11):1379 -1381.
13 Nagao T,Okura T,Miyoshi K,et al.Fibrillary glomerulonephritis associated with monoclonal gammopathy of undetermined significance showing lambda-type Bence Jones protein.Clin Exp Nephrol,2005,9(3):247-251.
14 Collins M,Navaneethan SD,Chung M,et al.Rituximab treatment of fibrillary glomerulonephritis. Am J Kidney Dis, 2008, 52(6):1158-1162.
15 Samaniego M,Nadasdy GM,Laszik Z,et al.Outcome of renal transplantation in fibrillary glomerulonephritis.Clin Nephrol,2001,55(2):159-166.
