遗传性C3肾炎
2017-05-09梁丹丹曾彩虹
梁丹丹 曾彩虹
·肾活检·
遗传性C3肾炎
梁丹丹 曾彩虹
青年男性,5岁起病,临床表现为中至大量蛋白尿,大量镜下血尿,肾功能缓慢减退,同时补体C3水平轻度下降。有肾脏疾病家族史。肾活检光镜初始改变为肾小球系膜增生性病变,重复肾活检见肾小球不典型膜增生性病变伴内皮下、系膜区大量嗜复红物沉积,免疫荧光以C3沉积为主,Ⅳ型胶原染色正常。电镜下肾小球系膜区、内皮下大量、基膜内节段电子致密物分布。基因测序未见补体相关基因突变。最终诊断为遗传性C3肾炎。
C3肾炎 肾活检
病例摘要
病史 24岁男性患者,因“尿检异常18年余,发现血清肌酐(SCr)升高2周”于2016-01-20入院。
患者1997年5岁时体检发现尿常规隐血++,肾功能正常,未予诊治。后多次复查尿常规示尿蛋白-,潜血+~++,未予治疗。2004年11月第一次至我科住院,查尿蛋白1.65 g/24h,尿沉渣红细胞80万/ml(多形型),SCr 46.9 μmol/L,双肾B超(LK/RK)114/111 mm、结构正常,耳电测听、双眼晶体检查等均未见异常,未查血补体水平,行肾活检光镜表现为肾小球系膜增生性病变伴系膜区、内皮下嗜复红物沉积,免疫荧光IgA+、C3++,弥漫分布于系膜区及血管袢,诊断为“IgA肾病”。予贝那普利、雷公藤多苷、新肾炎胶囊等治疗,后患者未定期随访。2011年~2013年间尿蛋白定量0.83~3.19 g/24h,尿沉渣红细胞83~290万/ml,多形型,SCr逐渐升至84.9 μmol/L。后未再规律随访及服药。2016-01-08我科门诊查SCr升至188.3 μmol/L,尿素氮(BUN)13 mmol/L,尿酸(UA)526 μmol/L。为求进一步诊治再次收住我科。病程中无水肿,无肉眼血尿,无听力或视力下降,无夜尿增多。精神尚可,体力、食欲正常,睡眠可,体重无明显变化,大便及排尿正常。
家族史 1个伯父25岁时发现肾脏病,外院肾活检诊断为膜增生性肾小球肾炎(MPGN),35岁(2002年)时进展至终末期肾病(ESRD),同年行肾移植术,2015年移植肾失功。父亲24岁时发现肾脏疾病,外院肾活检诊断为MPGN,34岁(2004年)时进展至ESRD,同年行肾移植术,2006年在我科行移植肾活检提示为C3肾炎,2010年移植肾失功开始于普通血透。祖母尿蛋白阳性,父亲的舅舅死于肾脏疾病(具体不详)(图1),母亲体健。既往史、个人史无特殊。
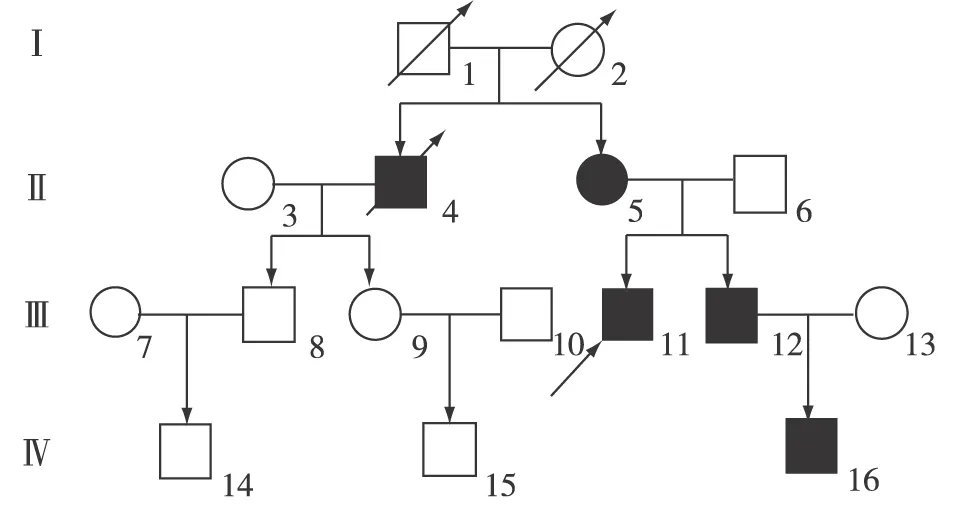
图1 患者家系图其中11号为患者伯父,为先证者;16号为患者
体格检查 体温36.5℃,脉搏103次/min,呼吸20次/min,血压177/102 mmHg,体质量指数18.9 kg/m2;神清,咽不红,扁桃体不大,心、肺、腹部未见明显异常,双下肢无水肿。肌力正常。
实验室检查
血常规 血红蛋白(Hb) 130 g/L,WBC 7.0×109/L,PLT 317×109/L。
血生化 白蛋白30.1 g/L,球蛋白18.0 g/L,BUN 24.1 mmol/L,SCr 196.2 μmol/L,UA 509 μmol/L,转氨酶正常,总胆固醇8.1 mmol/L,三酰甘油3.28 mmol/L,电解质无异常,钙2.09 mmol/L,磷1.54 mmol/L。
尿液 尿蛋白定量3.89 g/24h,尿沉渣红细胞105~200万/ml;尿C3 3.21 mg/L(正常值≤2.76 mg/L),α2-MG 3.27 mg/L(正常值≤2.87 mg/L),RBP 2.1 mg/L(正常值<0.5 mg/L),Lyso<0.5 mg/L,NAG 18.8 U/(g·Cr)[正常值≤16.5 U/(g·Cr)]。禁饮13h尿渗量363 mOsm/(kg·H2O)。
其他 ANA、A-dsDNA均阴性。IgG 5.41 g/L,IgA 1.280 g/L,IgM 0.904 g/L,IgE 72.5 IU/ml,ASO、RF正常,补体C3 0.69 gL,C4 0.151 g/L。传染病四项均阴性。C3肾炎因子、抗H因子抗体阴性,I因子、H因子水平正常。
影像学检查 双肾B超:左肾120 mm×47 mm×60 mm,右肾122 mm×53 mm×61 mm,肾皮质厚度不清,皮质回声稍增强,皮髓界限清楚,右肾内可见多个类圆形无回声区,较大的约10 mm×11 mm,部分囊周伴钙化,界清。左肾上极见一直径约3 mm的强回声光团,伴弱后声影。左肾内局部肾盂肾盏扩张。超声心动图大致正常。
首次肾活检(2004-11-17)
光镜 皮髓质肾组织2条。26个肾小球中2个球性废弃,1个节段硬化。余肾小球系膜区轻度增宽(图2A),基质增多,囊壁不规则增厚、节段分层。PASM-Masson:肾小球系膜区较多、内皮下少量嗜复红物沉积。肾小管间质病变轻,灶性肾小管上皮细胞细颗粒变性,偶见空泡变性,数处管腔内见红细胞管型。间质小灶性增宽,纤维化+,散在浸润细胞及泡沫细胞。间质动脉未见明确病变。
免疫荧光 冰冻切片荧光染色IgA+、C3+++(图2B、C),呈颗粒状弥漫分布于系膜区及血管袢,IgG、IgM、C4、C1q阴性。Ⅳ型胶原α3、α5链染色正常。
电镜 肾小球系膜区增宽,系膜基质增多,系膜区较多高密度电子致密物。肾小球毛细血管袢内皮细胞增生,胞质丰富,连拱状改变,袢腔内偶见髓样小体,节段内皮下疏松,基膜内皮下、基膜内节段高密度电子致密物沉积(图2D),基膜上皮侧未见电子致密物分布;肾小球毛细血管袢基膜厚约250~310 nm,节段基膜偏薄120~240 nm(图2E)。足细胞胞质丰富,胞质内较多溶酶体,足突节段融合。
小结:IgA肾病。
重复肾活检病理(2016-01-08)
光镜 皮质肾组织2条。42个肾小球中20个球性废弃,13个节段硬化。余肾小球系膜区未见明显增宽,毛细血管袢开放好,数个球袢皱缩、开放不佳、囊周纤维化,囊壁节段增厚。PASM-Masson:肾小球系膜区、内皮下大量嗜复红物沉积(图3A)。肾小管间质重度慢性病变合并急性病变,斑片状肾小管萎缩、基膜增厚,灶性肾小管上皮细胞刷状缘脱落,间质较多浸润细胞分布,以单个核细胞为主,灶性泡沫细胞。小叶间动脉内膜增厚、弹力层分层,动脉管壁增厚,见内皮下疏松。
免疫荧光 冰冻切片荧光染色IgM+、C3+++(图3B),呈颗粒状弥漫分布于系膜区及血管袢。IgG、IgA、C1q阴性。κ轻链+、λ轻链+,节段分布,呈颗粒状沉积于系膜区及血管袢。Ⅳ型胶原α3、α5链染色正常。
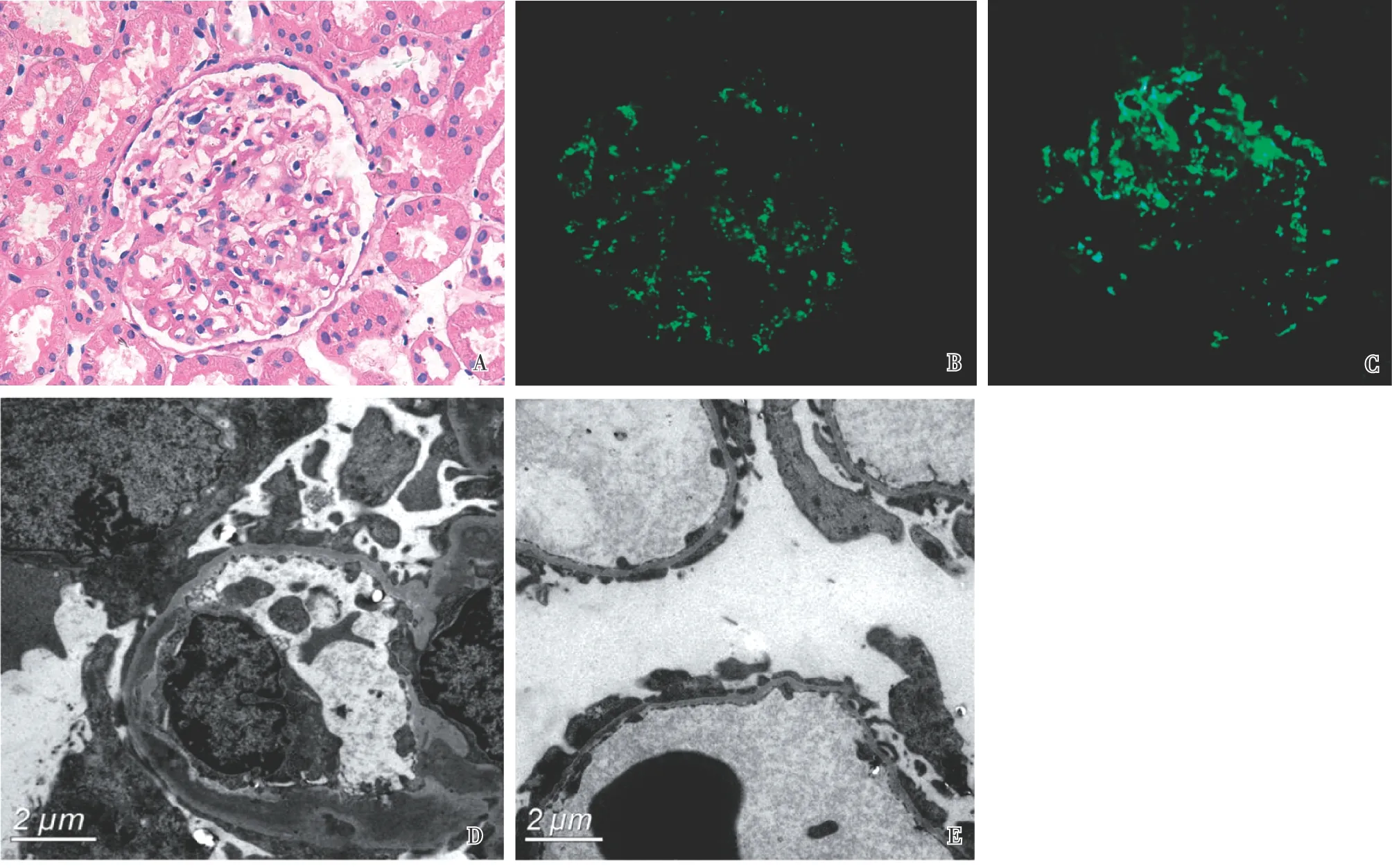
图2 第一次肾活检A:肾小球系膜区轻度增宽,系膜细胞及基质增多(PAS,×400);B:IgA+,呈颗粒状弥漫分布于系膜区及血管袢(IF,×400);C:C3+++,呈颗粒状弥漫分布于系膜区及血管袢(IF,×400);D:肾小球系膜区、内皮下见大量高密度的电子致密物分布,足细胞足突融合(EM);E:肾小球节段基膜偏薄(EM)

图3 重复肾活检A:肾小球系膜区增宽,系膜区、内皮下见大量嗜复红物沉积,间质小灶性单个核细胞浸润(Masson三色,×400);B:C3+++,呈颗粒状弥漫分布于系膜区及血管袢(IF,×400);C:肾小球毛细血管袢内皮下、基膜内、系膜区大量高密度的电子致密物分布,足细胞足突融合(EM);D:肾小球节段基膜外侧缘不规则,内皮下、基膜内见电子致密物分布,足细胞足突融合(EM)
电镜 肾小球系膜区明显增宽,系膜细胞增生,基质增多,系膜区大量密度不均的电子致密物沉积,高倍镜下未见特殊结构。肾小球毛细血管袢开放好,节段袢内皮细胞增生,节段袢内皮下疏松,袢腔内见单核细胞及中性粒细胞浸润,见细胞成份插入及新形成的基膜,基膜内皮下大量、基膜内节段电子致密物沉积,基膜上皮侧未见电子致密物分布(图3C);肾小球毛细血管袢基膜厚340~625 nm,节段基膜偏薄,厚约170~240 nm,节段基膜外侧缘不规则(图3D)。足细胞足突节段融合(30%~40%)。包囊壁及肾小管基膜未见电子致密物沉积。
小结:C3肾炎。
父亲移植肾活检(2006-12-28)
光镜 表现为肾小球中度系膜增生性病变(图4A)伴系膜区较多、内皮下少量嗜复红物沉积(图4B),肾小管间质轻度慢性病变,动脉未见明确病变。免疫荧光仅见C3++(图4C),呈颗粒状弥漫分布于系膜区及血管袢。电镜表现为肾小球系膜增生性病变伴系膜区大量、内皮下节段电子致密物分布(图4D),足细胞足突偶见融合。肾活检病理诊断为移植肾C3肾炎。

图4 患者父亲移植肾活检A:肾小球系膜区中度增宽,系膜细胞及基质增多,囊壁增厚分层(PAS,×400);B:肾小球系膜区较多、内皮下少量嗜复红物沉积(Masson三色,×400);C:C3++,呈颗粒状弥漫分布于系膜区及血管袢(IF,×400);D:肾小球系膜区、内皮下均见密度不均的电子致密物分布(EM)
讨 论
本例青年男性患者,幼年起病,临床表现为中至大量蛋白尿,大量镜下血尿,肾功能缓慢下降,补体C3水平轻度下降,肾脏病家族史阳性。肾脏病理光镜初始表现为肾小球系膜增生性病变,后进展为不典型膜增生性肾小球肾炎,免疫荧光以C3沉积为主,电镜下见肾小球内皮下、系膜区大量,基膜内少量电子致密物分布,电子致密物未见特殊超微结构。肾组织Ⅳ型胶原α3、α5链染色正常。
患者行第一次肾活检表现为肾小球系膜增生性病变,免疫荧光示IgA、C3沉积,因缺乏对C3肾炎的认识,当时主要鉴别原发性和继发性IgA肾病及以IgA沉积为主的感染后肾小球肾炎。IgA沉积为主的感染后肾小球肾炎好发于老年、糖尿病患者,光镜可表现为肾小球系膜增生性病变和毛细血管内增生性病变,电镜下除系膜区电子致密物外,尚见上皮侧“驼峰”形成,而该患者发病时年轻,慢性起病,发病前并无前驱感染史,电镜下亦未见“驼峰”,并不符合该诊断。同时未发现IgA肾病的继发病因如过敏性紫癜、强直性脊柱炎、银屑病、类风湿性关节炎、系统性红斑狼疮等,因此2004年诊断为IgA肾病。
重复肾活检时未见IgA沉积,仅表现为C3阳性,光镜呈不典型膜增生性肾小球肾炎,需进行以下鉴别诊断:感染(如乙型肝炎、丙型肝炎)、自身免疫性疾病(如系统性红斑狼疮)、肿瘤(如浆细胞性疾病)等导致的免疫球蛋白或免疫复合物相关的膜增生性肾小球肾炎,感染后肾小球肾炎,C3肾病等。根据免疫荧光仅少量或无IgG、IgA或IgM沉积,初步可排除单克隆或多克隆免疫球蛋白相关的膜增生性肾小球肾炎。感染后肾小球肾炎急性期光镜可见肾小球内大量中性粒细胞浸润等渗出性改变,即毛细血管内增生,伴或不伴毛细血管外增生,慢性期可仅表现为肾小球系膜增生性病变,电镜下可见肾小球毛细血管袢上皮侧“驼峰”状电子致密物沉积,亦可沉积于系膜区、内皮下,临床可出现ASO升高,该病大多数具有自限性,尿检、肾功能可恢复,补体水平多于8~12周内恢复正常,显然与该患者的慢性病程不符。因此,诊断C3肾病可能性很大。
C3肾病是一组表现为补体C3沉积于肾小球的疾病[1],包括致密物沉积病(DDD)和C3肾炎,两者鉴别主要依赖电镜,前者电子致密物分布于肾小球毛细血管袢内皮下、系膜区、上皮侧,可伴基膜内少量不连续沉积[2],而后者电子致密物主要分布于基膜内,呈“腊肠”样改变。其免疫荧光特点是仅见C3沉积或以C3沉积为主,即C3的强度比其他免疫球蛋白及补体高出2个等级[3]。光镜下C3肾病可表现为肾小球膜增生性病变、弥漫增生性病变、系膜增生性病变,甚至坏死性新月体肾炎[4]。C3肾病患者诊断时的中位年龄是21岁,临床上一些DDD患者会出现获得性部分脂质营养不良和脉络膜玻璃膜疣[5]。30%~50%C3肾病患者在10年内进展至ESRD,50%~75%在移植后出现C3肾病复发并可导致移植肾失功[6-8]。Zand等[9]报道C3肾病致ESRD的患者在肾移植后66.7%出现复发,复发中位时间是28个月,其中50%患者出现移植肾失功。该患者免疫荧光以C3沉积为主,虽第一次肾活检见少量IgA沉积,重复活检并未见IgA阳性,且光镜和电镜下沉积物范围广,主要分布于系膜区和内皮下,少数未分层的基膜致密层内见电子致密物沉积,电镜下电子致密物无特殊结构,无上皮侧“驼峰”状沉积物,未见基膜内“腊肠”样高密度电子致密物,不同于常见的IgA肾病,而在C3肾小球肾炎患者中常出现,因此最终诊断为C3肾炎。
从病因上看,C3肾病发病主要是由于补体旁路途径先天性或后天性调节异常所致,包括各种补体及相关调节蛋白基因突变和影响补体活性的抗体。目前有各种基因突变引起遗传性DDD和C3肾炎的报道[10-14];体内各种调节蛋白的抗体亦能致病,如C3肾炎因子[15]、C4肾炎因子[16]、抗B因子抗体、抗C3b抗体[17]等;尚有单克隆免疫球蛋白血症导致C3肾炎的报道[18]。该患者父亲家族中多人患肾脏疾病,并呈常染色体显性方式遗传,父亲和伯伯均于30余岁时进展至ESRD,二者曾在外院行自体肾肾活检诊断为MPGN,但年代已久,具体资料不详,而父亲在我院的移植肾活检亦提示为C3肾炎,因此高度考虑为家族遗传性C3肾炎。进而采用二代测序外显子序列捕获技术对本例患者进行基因测序,并未发现补体相关基因突变,却检测出COL4A4基因缺失突变,COL4A4基因c.1505(E22)delC,通过对其父母进行验证证实该杂合突变为母亲来源,但母亲及其家族中并无人检出尿检异常。同时,该患者临床眼、耳检查未见异常,电镜下虽见肾小球基膜节段偏薄,基膜外侧缘不规则,但无典型Alport综合征的“花篮”状改变,肾组织Ⅳ型胶原染色亦正常,进一步证实COL4A4基因杂合突变导致肾功能恶化的证据不足。针对该患者的补体相关检查结果显示I因子、H因子正常,C3肾炎因子、抗H因子抗体阴性,免疫固定电泳亦未查及单克隆免疫球蛋白条带。基因测序虽未检测到补体及相关调节蛋白的基因突变,但由于该检查为外显子测序,存在一定局限性,结合患者的家族史及父亲的移植肾活检病理,仍需高度考虑遗传性C3肾炎。
该患者的诊断过程存在一些波折。第一次肾活检光镜表现为肾小球系膜增生性病变,免疫荧光见IgA、C3沉积于肾小球系膜区及血管袢,虽然C3强于IgA,电镜下肾小球内皮下、系膜区,甚至节段基膜内均见电子致密物分布,但2004年对C3肾炎的认识不足,当时诊断为IgA肾病。目前来看,第一次肾活检时C3比IgA的强度高2个等级,且重复肾活检中IgA沉积消失,但光镜和电镜下肾组织的沉积物并无减少,反而更广泛、弥漫,均提示C3沉积在肾脏致病过程中的主要作用,因此对初次诊断为IgA肾病应存质疑。另外,近些年来对于C3肾炎发病机制及病理改变的研究都有了很大进展,患者重复肾活检为典型的C3肾炎表现,即免疫荧光以C3沉积为主,肾小球表现为不典型膜增生性病变,系膜区、内皮下,同时少量基膜内均见大量沉积物,最终确诊为C3肾炎,结合其家族史,诊断为遗传性C3肾炎。目前针对该类疾病的治疗仅基于一些单中心研究,包括抗补体治疗、免疫抑制剂及一些减少蛋白尿的对症治疗,效果欠佳,本例患者在第二次肾活检后随访10个月时进展至ESRD。
小结:本文报告1例24岁男性患者临床表现为尿检异常伴肾功能缓慢下降,补体C3轻度降低,肾脏病家族史阳性,并呈常染色体显性遗传,经两次重复肾活检最终诊断为遗传性C3肾炎。目前针对该病的治疗方法和效果尚缺乏多中心大样本的临床数据支持,有待进一步研究。
1 Barbour TD,Ruseva MM,Pickering MC.Update on C3 glomerulopathy.Nephrol Dial Transplant,2016,31(5):717-725.
2 Sethi S,Fervenza FC.Pathology of renal diseases associated with dysfunction of the alternative pathway of complement:C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS).Semin Thromb Hemost,2014,40(4):416-421.
3 Hou J,Markowitz GS,Bomback AS,et al.Toward a working definition of C3 glomerulopathy by immunofluorescence.Kidney Int,2014,85(2):450-456.
4 Pickering MC,D'Agati VD,Nester CM,et al.C3 glomerulopathy:consensus report.Kidney Int,2013,84(6):1079-1089.
5 Servais A,No⊇l LH,Frémeaux-Bacchi V,et al.C3 glomerulopathy.Contrib Nephrol,2013,181:185-93.
6 Medjeral-Thomas NR,O'Shaughnessy MM,O'Regan JA,et al.C3 glomerulopathy:clinicopathologic features and predictors of outcome.Clin J Am Soc Nephrol,2014,9(1):46-53.
7 Lu DF,Moon M,Lanning LD,et al.Clinical features and outcomes of 98 children and adults with dense deposit disease.Pediatr Nephrol,2012,27(5):773-781.
8 Servais A,No⊇l LH,Roumenina LT,et al.Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies.Kidney Int,2012,82(4):454-464.
9 Zand L,Lorenz EC,Cosio FG,et al.Clinical findings,pathology,and outcomes of C3GN after kidney transplantation.J Am Soc Nephrol,2014,25(5):1110-1117.
10 Gale DP,de Jorge EG,Cook HT,et al.Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis.Lancet,2010,376(9743):794-801.
11 Athanasiou Y,Voskarides K,Gale DP,et al.Familial C3 glomerulopathy associated with CFHR5 mutations:clinical characteristics of 91 patients in 16 pedigrees.Clin J Am Soc Nephrol,2011,6(6):1436-1446.
12 Malik TH,Lavin PJ,Goicoechea de Jorge E,et al.A hybrid CFHR3-1 gene causes familial C3 glomerulopathy.J Am Soc Nephrol,2012,23(7):1155-1160.
13 Tortajada A,Yébenes H,Abarrategui-Garrido C,et al.C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation.J Clin Invest,2013,123(6):2434-2446.
14 Medjeral-Thomas N,Malik TH,Patel MP,et al.A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry.Kidney Int,2014,85(4):933-937.
15 Spitzer RE,Stitzel AE.On the origin and control of C3NeF.In vivo,1988,2(1):79-81.
16 Ohi H,Yasugi T.Occurrence of C3 nephritic factor and C4 nephritic factor in membranoproliferative glomerulonephritis (MPGN).Clin Exp Immunol,1994,95(2):316-321.
17 Chen Q,Müller D,Rudolph B,et al.Combined C3b and factor B autoantibodies and MPGN type II.N Engl J Med,2011,365(24):2340-2342.
18 Lloyd IE,Gallan A,Huston HK,et al.C3 glomerulopathy in adults:a distinct patient subset showing frequent association with monoclonal gammopathy and poor renal outcome.Clin Kidney J,2016,9(6):794-799.
(本文编辑 律 舟)
Familial C3 glomerulonephritis
LIANGDandan,ZENGCaihong
NationalClinicalResearchCenterofKidneyDiseases,JinlingHospital,NanjingUniversitySchoolofMedicine,Nanjing210016,China
A young male with a family history of kidney disease exhibited microscopic hematuria from 5 years old. He gradually developed proteinuria, renal insufficiency and mildly decreased C3 level. A kidney biopsy was performed and light microscopic findings demonstrated mesangioproliferative glomerulonephritis. Repeat kidney biopsy showed atypical membranoproliferative glomerulonephritis. C3 dominant staining and normal for α3(Ⅳ) and α5 (Ⅳ) collagen staining were seen by immunofluorescence. On electron microscopy, it showed massive mesangial and subendothelial and segmental discontinuous intramembranous electron dense deposits. Complement mutations weren't detected by genetic sequencing. Finally, the patient was diagnosed with familial C3 glomerulonephritis.
C3 glomerulonephritis renal biopsy
10.3969/cndt.j.issn.1006-298X.2017.02.019
南京军区南京总医院肾脏科 国家肾脏疾病临床医学研究中心 全军肾脏病研究所(南京,210016)
2017-02-14
