常染色体显性遗传腓骨肌萎缩症的临床与基因突变特点
2011-08-11宋福聪王相斌冯文霞胡志强唐北沙
郭 鹏, 宋福聪, 王相斌, 冯文霞, 胡志强, 唐北沙, 夏 昆
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)是一组最常见的具有高度临床和遗传异质性的周围神经单基因遗传病,遗传方式可为常染色体显性遗传、常染色体隐性遗传和X连锁遗传。患病率约为1/2500,主要特征为肢体远端进行性肌无力和肌萎缩伴腱反射减退或消失[1]。根据电生理和病理特征,CMT可分为CMT1(脱髓鞘型)和CMT2(轴索型)。
目前至少已发现39个不同的CMT致病基因位点,其中27个基因相继被克隆,其中常染色体显性遗传CMT致病基因主要有PMP22、MPZ、SIMPLE、EGR2、KIF1Bβ、MFN2、RAB7、GARS、NEFL、Hsp27及 Hsp22 基因(http://molgen-www.ua.ac.be/CMTMutations)。
我们应用聚合酶链反应(PCR)、实时荧光定量PCR法、变性高效液相色谱(DHPLC)结合DNA直接测序和限制性内切酶酶切技术,对43个常染色体显性遗传CMT家系进行PMP22的大片段重复突变和 PMP22、MPZ、SIMPLE、EGR2、MFN2、RAB7、NE-FL、Hsp27及Hsp22基因突变分析,并分析其临床表现、电生理和病理特点。
1 对象与方法
1.1 对象 43个常染色体显性遗传CMT家系来源于邯郸市中心医院神经内科、中南大学湘雅医院神经内科及医学遗传学国家重点实验室所收集的病例。家系先证者均由两名神经内科专科医生进行详细的病史询问和体格检查,符合Harding于1980 年制定的诊断标准[2]。
1.2 周围神经电生理检查 神经电生理检查包括正中神经、尺神经、胫神经和腓总神经运动和感觉神经传导速度检查。将运动神经传导速度减慢程度分为重度降低(<10m/s)、显著降低(10m~38m/s)、和减慢不明显(>38m/s)[3]。
1.3 神经活检 常规皮肤消毒,局麻,于外踝和跟腱正中切开皮肤,在皮下组织内寻找腓神经,切取腓神经约2cm,缝合,用4%多聚甲醛固定,石蜡包埋,进行常规的HE染色、甲苯胺蓝染色、半薄切片检查,光镜观察。
1.4 基因突变分析 采用实时荧光定量PCR法对每个家系先证者进行PMP22基因大片段重复突变检测,对MFN2采用DHPLC、DNA直接测序和限制性内切酶酶切检测,对 PMP22、MPZ、SIMPLE、EGR2、RAB7、NEFL、Hsp27及 Hsp22进行聚合酶链反应(PCR)、DNA直接测序和限制性内切酶酶切检测。
2 结果
2.1 临床特点 43个腓骨肌萎缩症家系共106例患者中男性65例,女性41例,发病年龄1~60岁,平均发病年龄19.1岁。首发症状为小腿肌萎缩伴无力者90例,手、足同时萎缩者5例,手肌萎缩者1例。体格检查:105例均存在不同程度的双下肢萎缩和肌力下降,严重者双膝关节以下肌肉极度萎缩呈鹤腿状,双上肢无力伴萎缩者41例;双上肢肌张力下降30例,双下肢肌张力下降88例;双上肢远端感觉减退或缺失30例,双下肢远端浅感觉减退或缺失65例;22例踝反射消失,70例膝、踝反射减退;弓形足29例,爪形手6例,足内翻3例,脊柱侧弯畸形3例。
2.2 周围神经电生理改变 34例进行了肌电图检查均可见巨大运动单元电位、纤颤电位和正锐波,表现为广泛神经源性损害。16例患者胫神经和腓神经传导速度重度降低(<10m/s),8例患者下肢神经传导速度显著降低(<38m/s),另外10例传导速度减慢不明显(>38m/s)。24例患者上肢正中神经和尺神经传导速度显著降低(<38m/s),为CMT1型;另外10例传导速度减慢不明显(>38m/s),为 CMT2型。
2.3 神经活检改变 26例患者行腓神经活检光镜检查,发现CMT1型16例,7例髓鞘轻度脱失,6例中到重度脱失,7例髓鞘增厚,4例呈“洋葱球”样改变,均无轴索变性;CMT2型10例,均出现慢性的髓鞘化轴索萎缩、缺失、再生等轴索变性表现,3例同时有髓鞘轻度脱失,2例有髓鞘增厚,1例可见“洋葱球”样改变。
2.4 基因突变分析 在18个家系共55例患者中发现PMP22基因大片段重复突变[4];在1个家系共4例患者中发现MPZ基因突变 Thr124Met[4],在2个家系共2例患者中发现MFN2基因突变Arg94Gln和Cys132Thr[5];在1个家系共18例患者中发现Hsp27基因突变Arg127Trp[6];在1个家系共4例患者中发现Hsp22基因突变Lys141Asn[7],而在所有患者中未发现 SIMPLE、EGR2、RAB7、NEFL基因突变[8]。基因型与临床特点(见表1)。
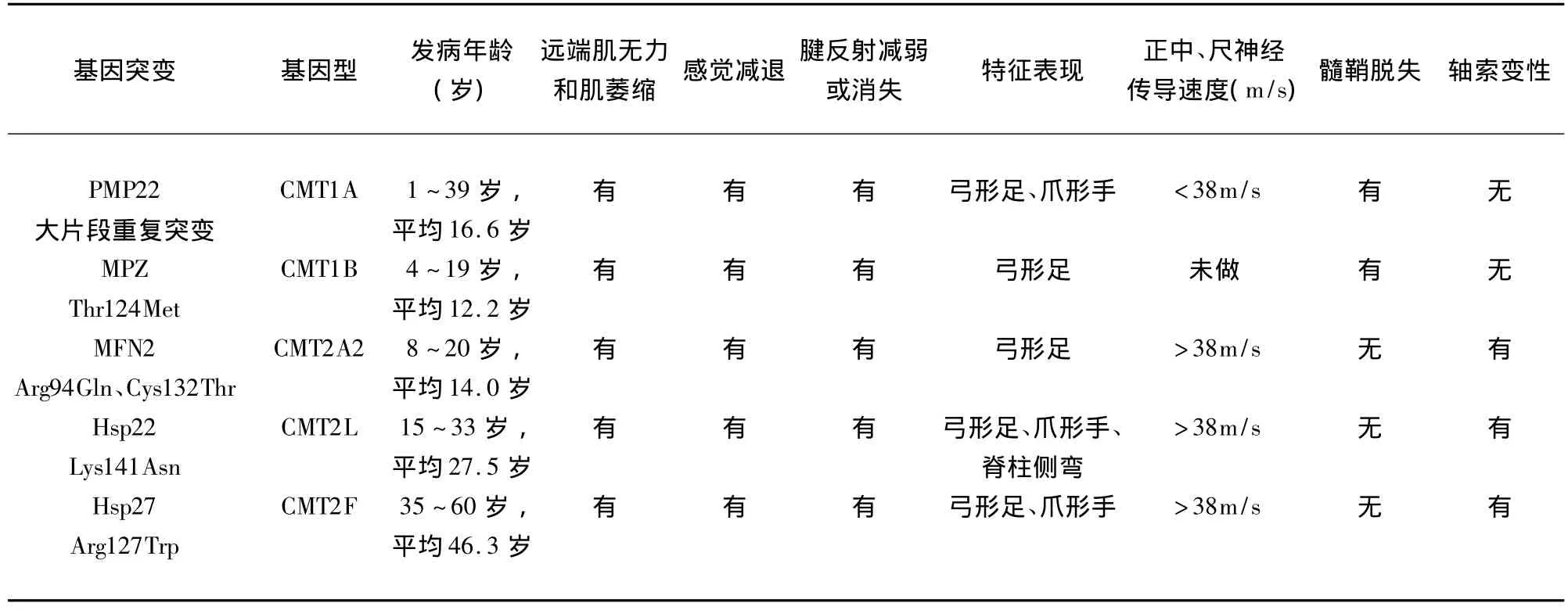
表1 CMT基因型与临床特点
3 讨论
本组常染色体显性遗传CMT患者具有以下临床特征:在儿童期、青年期或青年晚期发病,个别中老年起病,起病隐袭,进展缓慢;临床表现多见起始于下肌远端的无力萎缩,可累及上肢,伴感觉减退或缺失。神经系统检查可见小腿肌肉明显萎缩,呈“鹤腿样”表现。膝、踝反射减低或消失,部分有弓形足、足内翻、爪形手、脊柱侧突等畸形。根据周围神经电生理和病理特点可将CMT分为两型:CMT1(脱髓鞘型),其正中神经运动传导速度<38m/s,神经活检示广泛的节段性脱髓鞘和髓鞘增生形成洋葱球样结构;CMT2(轴索型),其正中神经运动传导速度正常或轻度减慢(>38m/s),神经活检示轴索变性。根据基因突变分析结果将本组患者基因型分为CMT 1A、CMT 1B、CMT2A2、CMT2F和 CMT2L。
本组CMT1临床特点主要为:(1)发病年龄较小;(2)特征性临床表现是肢体远端无力和肌萎缩;(3)以弓形足为主的骨骼畸形常见,严重的骨骼畸形如脊柱侧凸罕见;(4)感觉障碍较常见;(5)正中神经运动传导速度<38m/s;(6)神经活检发现髓鞘脱失。CMT1A占CMT1的70% ~80%,PMP22大片段重复突变导致编码蛋白的过度表达是导致CMT1A的最常见因素。PMP22基因编码周围神经髓鞘蛋白22,在周围神经中有高度表达,其功能可能为维持髓鞘结构的完整性、调节细胞周期及作为粘附分子。过度表达的PMP22不能进行正常的细胞内转移而积聚在Golgi复合体中,影响雪旺氏细胞的正常增生和分化;也可能由于PMP22与髓鞘蛋白零(myelin protein zero,MPZ)组成一个复合体以保持髓鞘的稳定,在CMT1患者中PMP22/MPZ的比例升高,破坏了髓鞘的稳定性[9]。CMT1B占 CMT1的5%~10%,与MPZ基因突变有关。MPZ编码周围神经髓磷脂的主要结构蛋白即髓鞘蛋白零蛋白,功能是通过与邻近的髓鞘板层相连保持髓磷脂的稳定,并对髓磷脂的致密化有一定作用。突变可影响MPZ蛋白的所有成分,导致髓磷脂附着减少[10]。
本组CMT2临床特点主要为:(1)发病年龄比CMT1迟,多在15岁后发病,仅有1例8岁发病;(2)运动系统受累较感觉系统更明显;(3)骨骼畸形如脊柱侧弯较CMT1常见;(4)正中神经运动传导速度>38m/s;(5)神经活检发现轴索变性。CMT2A2在CMT2中占20%以上,是目前最常见的CMT2亚型,与MNF2基因突变有关。MFN2通过介导线粒体融合而调节线粒体的网络结构。MFN2在各种组织中广泛表达,但其基因突变仅导致轴索型周围神经遗传病,这可能和周围神经长轴突结构、维持长距离轴浆运输更需要依赖完整线粒体网络提供能量有关[11]。Hsp27、Hsp22 基因突变可以分别引起CMT2F和CMT2L,它们所编码的蛋白同属于热休克蛋白超家族,该超家族是细胞信号传导通路的一部分,具有分子伴侣样功能,它们可以通过抗细胞调亡作用提高细胞在应激状态下的生存能力[12]。Hsp22基因突变可能会影响轴索运输;而突变型Hsp27蛋白可降低神经元细胞的生存能力,并损害神经丝的组装[13],后者对维持神经元轴突细胞骨架和轴突转运起关键作用。在所有患者中未发现 SIMPLE、EGR2、RAB7、NEFL基因突变,说明上述基因突变在中国人中罕见。
长期以来,CMT的诊断依据的是临床、电生理和病理资料,但是由于CMT具有明显的临床异质性,不同基因突变所致的CMT患者从表型上很难区分,而且并非所有患者都有明显的临床或电生理改变,另外,神经活检的损伤比较大,患者接受程度低。相比之下基因诊断的准确性高,无损伤,并且在病程早期即可做出诊断确定基因型,值得广泛应用于临床,特别是有家族史的患者或高危亲属,对优生优育和患者的早期治疗有重要意义。
[1]Berger P,Young P,Suter U.Molecular cell biology of Charcot-Marie-Tooth disease[J].Neurogenetics,2002,4(1):1-15.
[2]Harding AE,Thomas PK.The clinical features of hereditary motor and sensory neutopathy typesⅠandⅡ[J].Brain,1980,103(2):258-280.
[3]Pareyson D,Scaioli V,Laura M.Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease[J].Neuromolecular Med,2006,8(1):3-22.
[4]张付峰,唐北沙,赵国华,等.腓骨肌萎缩症致病基因突变特点的研究[J].中华医学杂志,2005,85(26):1809-1812.
[5]张如旭,付 敏,资晓宏,等.中国人腓骨肌萎缩症线粒体融合蛋白2 基因突变分析[J].中华医学杂志,2009,89(47):3324-3327.
[6]Tang BS,Liu X,Zhao GH,et al.Mutation analysis of the small heatshock protein 27 gene in Chinese patients with Charcot-Marie-Tooth disease[J].Arch Neurol,2005,62(8):1201-1207.
[7]Tang BS,Zhao GH,Luo W,et al.Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L[J].Hum Genet,2005,116(3):222-224.
[8]张如旭,郭 鹏,任志军,等.LITAF、RAB7、LMNA和MTMR2基因在中国人腓骨肌萎缩患者的突变分析[J].遗传,2010,32(8):817-823.
[9]Nobbio L,Vigo T,Abbruzzese M,et al.Impairment of PMP22 transgenic Schwann cells differentiation in culture:implications for Charcot-Marie-Tooth type 1A disease[J].Neurobiol Dis,2004,16(1):263-273.
[10]Runker AE,Kobsar I,Fink T,et al.Pathology of a mouse mutation in peripheral myelin protein P0 is characteristic of a severe and early onset form of human Charcot-Marie-Tooth type 1B disorder[J].Cell Biol,2004,165(4):565-573.
[11]Kijima K,Numakura C,Izumino H,et al.Mitochondrial GTpase mitofusin 2 mutation in Charcot-marie-tooth neuropathy type 2A [J].Hum Genet,2005,116(1):23-27.
[12]Benndorf R,Welsh MJ.Shocking degeneration[J].Nat Genet,2004,36(6):547-548.
[13]Evgrafov OV,Mersiyanova I,Irobi J,et al.Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy[J].Nat Genet,2004,36(6):602-606.
