H2S在五边形BCN上的吸附与解离的第一性原理计算研究
2024-02-01史金磊时俊仙王丽丽
史金磊, 时俊仙, 付 龙, 赵 高, 王丽丽
(郑州师范学院 物理与电子工程学院 郑州市低维微纳米材料重点实验室,郑州 450044)
1 引 言
H2S长期以来被认为是一种极其危险和具有腐蚀性的气体,对人类生活环境与身体健康产生严重危害,因此迫切需要一些新材料及净化技术来探测这些有害气体分子并予以过滤和消除,这对大气污染防治具有重要的意义.
通常工业上通过Clause工艺来处理H2S,即H2S+1/2O2→H2O+S. 然而,该过程会消耗大量热量,并且其副产物(如 SOx)会带来严重的环境问题[1]. 为此,迫切需要探索一种绿色且温和的方法来消除H2S气体. 近来,光催化技术已被证明是一种非常有前景的光分解 H2S 的方法,即H2S→H2+S[2-4],该过程不仅能够有效地降低能量损耗,同时还可以产生清洁的氢能. 然而,迄今为止,H2S 在光催化剂表面的吸附和解离机理尚未阐明. 此外,实验上可制备的金红石相TiO2(110)表面,被用于研究H2S分子在其表面可能的吸附与解离机制,H2S+TiO2(110)→S-TiO2(110)+H2O[5]. 然而,在完整的金红石相TiO2(110)表面,其H2S分子的解离势垒较大同样需要消耗热量.
此外,二维材料具有较大的表面积、具有奇异的物理化学性质及容易被外场调控等优点而备受关注,并且在气敏方面具有广泛的应用前景[6,7]. 例如,Mn掺杂石墨烯可以显著提高H2S在衬底上的吸附能及界面处的电荷转移[8]. Ni掺杂的InN对H2S分子具有较强的吸附性能[9]. 此外,Aghaei等人研究发现硅烯纳米带可用于H2S的气敏探测[10]. 最近研究发现,对MoS2引入异原子掺杂可以大大提高该材料对SO2及H2S分子的吸附能及表面处的电荷转移,并且过渡金属原子掺杂的MoS2单层可以用于SO2及H2S分子的净化消除[11-12]. 然而目前理论报道都基于原子掺杂,而实验上实现对材料精准掺杂仍然面临挑战. 因此,亟需探索新的材料体系来消除H2S气体.
最近被广泛报道的五边形石墨烯基材料[13-14],如五边形石墨烯[13,15]、五边形CB2[16]、CN2[17],BN2[18]与BCN[19]等,具有出色的力学和动力学稳定性,其新奇的电子性能在气敏及催化方面都有潜在应用前景. 然而到目前为止,仅有少量的报道研究小分子气体在五边形石墨烯基材料上的吸附性能. 例如,Cheng等人研究发现五边形石墨烯单层对NOx(x=1,2)气体具有较好的吸附性能在气敏元件具有应用潜力[20-22]. 然而,目前尚未有文献报道该材料体系对H2S分子的吸附与解离研究,其表面活性位点、反应中间体和可能的反应路线等相关问题都值得深入研究.
本文采用基于密度泛函理论的第一性原理计算方法,研究了H2S分子在五边形BCN(penta-BCN)上的吸附与解离过程. 该研究结果为penta-BCN对H2S分子的吸附解离机制提供理论借鉴.
2 计算方法与模型
计算基于密度泛函理论(DFT)的VASP程序包[23]. 采用全电子的投影缀加平面波方法(PAW)描述芯电子与价电子间的相互作用,对于交换-关联相互作用,采取基于广义梯度近似的PBE泛函[24],计算中电子波函数展开所采用的平面波矢的截断能选取500 eV. 结构弛豫及电子结构计算采用Monkhorst-Pack型网格,k点分别为3×3×1 及 7×7×1. 模型采用3 × 3 超胞(包含54个原子)的penta-BCN,表面沿法线方向设置 20 Å真空层以避免超胞间相互作用. 扩散能垒计算采用CINEB算法搜索过渡态,研究过渡金属原子在衬底表面的热力学稳定性.
3 结果与讨论
3.1 Penta-BCN结构模型与电子结构
如图1(a)所示,优化的penta-BCN的晶格常数a=3.67,b=3.63 Å,层厚度为h=1.32 Å. Penta-BCN原胞结构如1(a)中虚线所示,其原胞结构共有6个原子,B∶C∶N原子的比例为1∶1∶1. 结构中B-N,C-N 和 C-B键长分别1.41 Å,1.51 Å和1.62 Å. 此外,如图1(c)所示,我们还进一步研究了penta-BCN的电子能带结构,从图中可以看出penta-BCN是直接带隙半导体,所计算的带隙值约为1.70 eV,与之前报道相符[19].
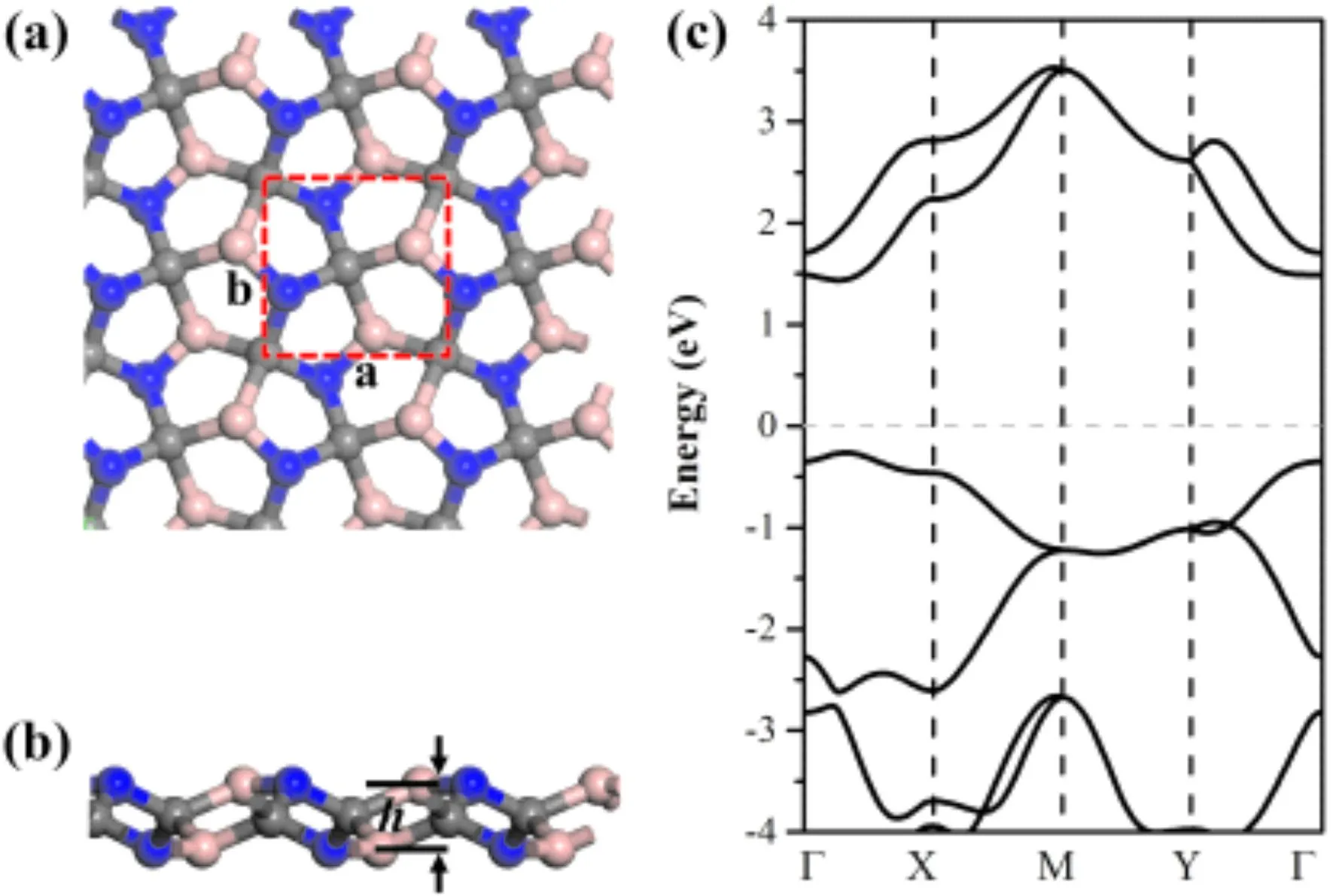
图1 (a)Penta-BCN结构3 × 3超胞模型;(b)Penta-BCN结构3 × 3超胞侧视图;(c)Penta-BCN电子能带结构.Fig. 1 (a)Penta-BCN 3 × 3 supercell. (b)Side view of the penta-BCN structures. (c)Band structures of penta-BCN.
3.2 H2S分子的吸附及分解
为了寻找H2S在衬底penta-BCN上的最稳定吸附构型,计算中我们采取将H2S气体分子放置于衬底上不同角度和各种可能的吸附位点. H2S在penta-BCN表面最稳定的吸附构型如图2(a)所示,其吸附能为0.537 eV. H2S中S原子局域在衬底表面的B原子的顶位,形成稳定的B-S键,键长为2.06 Å. 其中H2S中HS键的键长与H-S-H的键角分别为1.36 Å,1.37 Å和91.4°.其中HS键长(1.37 Å)与自由H2S气体分子(1.35 Å)相比显然拉伸,且氢原子指向近邻的N原子,也暗示着H2S在衬底上较易进行脱氢反应.
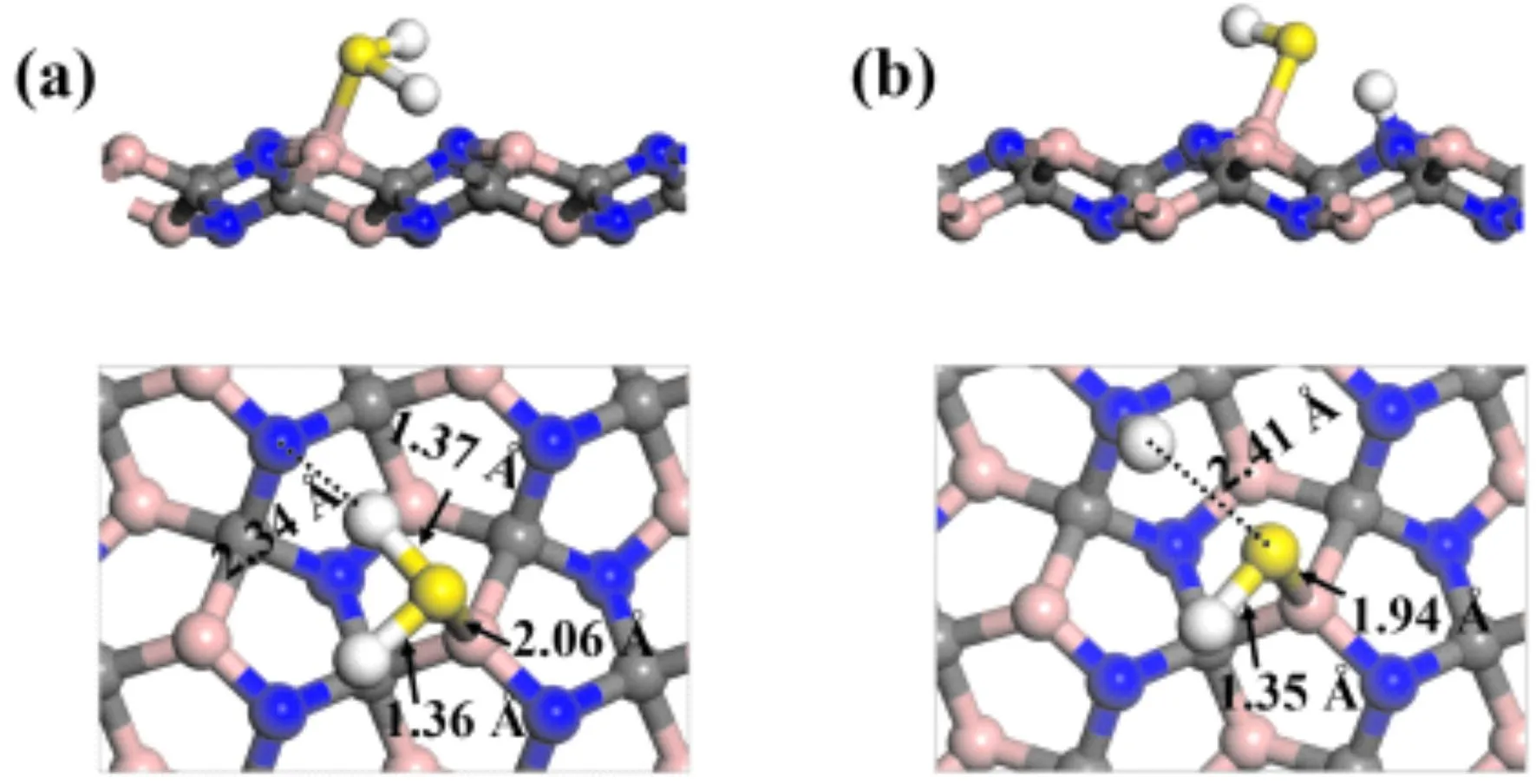
图2 (a)H2S在penta-BCN衬底上的吸附结构;(b)H2S在penta-BCN衬底上的解离吸附结构Fig. 2 (a)Adsorption structure of H2S on penta-BCN. (b)Dissociated configurations of H2S (H/HS)on the penta-BCN.
为了进一步阐明H2S在penta-BCN上的解离机制,我们在图3中给出了H2S分子在penta-BCN表面初步反应能量最低的反应路径. 从图中我们可以看出,吸附在衬底上的H2S分子首先经过脱氢反应,即H2S→SH + H,如图2(b)所示,解离后的产物在表面形成SH基,此时HS键的键长为1.35 Å. 与此同时解离的H原子扩散至近邻的N原子,形成稳定的N-H键,键长为1.05 Å. 值得注意的是,H2S分子在该反应过程中仅需克服0.208 eV的能量势垒,并且伴随释放 0.129 eV的能量,其中过渡态(如图3中TS)中HS键的键长分别为1.36 Å和1.64 Å. 基于此,我们还进一步考虑了HS在衬底上的进一步解离反应,即HS→S+H,然而,计算结果表明,HS分解后的体系能量比解离前体系能量升高约2.042 eV,也意味着HS在penta-BCN上很难进一步解离.
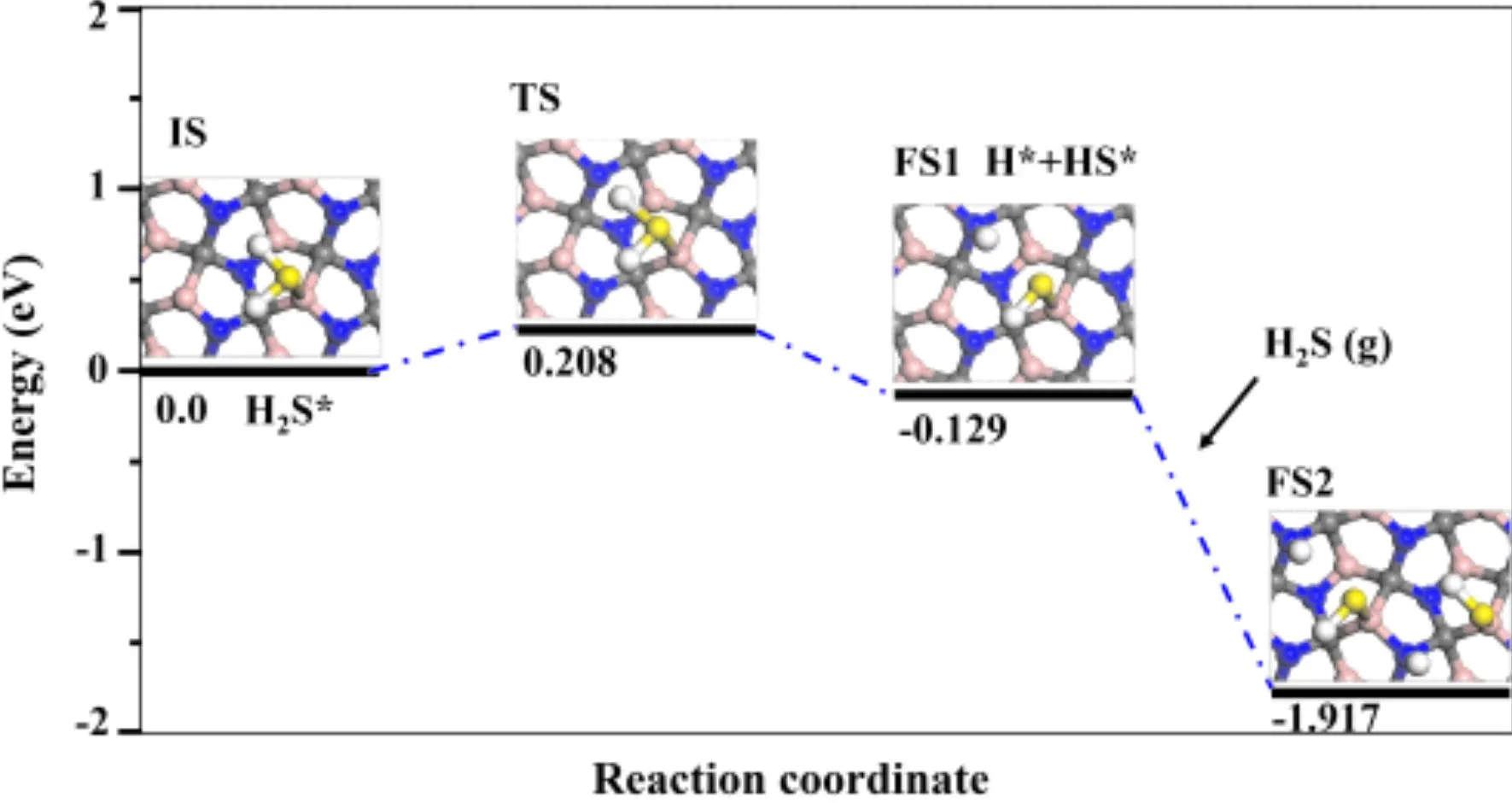
图3 H2S在penta-BCN上分解的势能分布示意图Fig. 3 Schematic potential energy profiles for the decomposition of H2S on the penta-BCN.
此外,深入研究发现,对于H2S在penta-BCN上初步解离后的体系(图(3)FS1),却可以直接解离H2S分子,在衬底形成稳定的SH/H表面结构并且伴随释放 1.788 eV的能量. 所以,从计算结果可以看出,H2S在penta-BCN上很容易解离,并且解离后的表面结构促进了后续H2S分子的分解.
3.3 H2S、H/HS 在penta-BCN上电子能带分析
为了深入了解H2S在penta-BCN上解离后结构促进后续H2S分子的解离机理,我们系统分析了penta-BCN、H2S吸附后及H2S解离体系的分波态密度图,如图4所示. 对于penta-BCN,从其分波态密度图(图4(a))可以看出,其价带顶主要由B原子和N原子贡献,其导带底主要由B原子贡献. 当H2S分子吸附时,(图4(b))可以看出体系的导带底下移,价带顶B原子与N原子及S原子间的局域的p轨道杂化,表明H2S吸附时在衬底界面处引起了界面间的电荷转移. 当H2S经历脱氢反应,解离成HS/H表面结构时,如图4(c)所示,其费米面附近形成更加局域的杂化轨道,导带底主要由B原子的p轨道贡献,价带顶则主要由N的p轨道贡献,并且其带隙与H2S吸附前相比,大大降低,这也表明HS/H表面结构导致的显著的带隙降低将极大提高表面催化活性,促进电荷转移及增强界面相互作用.

图4 (a)Penta-BCN,(b)H2S吸附后及(c)H2S分解后的分波态密度图Fig. 4 (a)The PDOS of penta-BCN substrate. (b)PDOS of H2S adsorption on penta-BCN,and (c)PDOS of dissociated SH/H on penta-BCN.
4 结 论
本文采用基于密度泛函理论的第一性原理计算方法,研究了H2S分子在penta-BCN上的吸附与解离过程. 研究结果表明,H2S分子在penta-BCN表面极易解离,其初步分解势垒仅为0.208 eV,并形成稳定的HS/H产物. 然而,H2S分子初步解离后的penta-BCN表面却可直接分解后续吸附的H2S分子,深入研究发现这是由于初步解离的HS/H产物导致带隙显著降低极大提高表面催化活性,促进电荷转移及增强界面相互作用,从而促进了后续H2S分子的解离. 该研究结果不仅为penta-BCN对H2S分子的吸附解离机制提供理论借鉴,并且提出penta-BCN材料可作为净化有害气体H2S的理想候选者.
