6-巯基-5-三唑并[4,3-b]-s-四嗪(MTT)的密度泛函理论研究
2024-02-01任黎英邵长斌陈玉锋
赵 宁, 陈 慧, 任黎英, 邵长斌, 陈玉锋
(牡丹江师范学院 化学化工学院 黑龙江省光电功能材料重点实验室, 牡丹江 157011)
1 引 言
6-巯基-5-三唑并[4,3-b]-s-四嗪(6-mercapto-5-triazolo[4,3-b]-s-tetrazine,MTT),是甲醛与4-氨基-5肼基-3-巯基-1,2,4-三唑(4-amino-3-hydrazine-5-mercapto-1,2,4-triazole,AHMT)在碱性环境下发生衍生化反应生成的产物之一. 甲醛作为一种生活中常见的污染物,也是国际癌症研究机构公布的致癌物清单中的一类致癌物[1],国标法检测甲醛[2,3]常使用AHMT分光光度法[4]. 朱等基于甲醛在室温下与AHMT进行氧化还原反应的原理,开发了一种高效便捷检测甲醛的 SERS 传感器,采用表面增强拉曼光谱的方法测定了甲醛;马等基于衍生化反应的超灵敏甲醛 SERS 传感器,超灵敏检测了食物和环境水中的痕量甲醛[5-8]. 但对于MTT分子的结构、分子振动方式、分子轨道等微观结构的理论研究未见报道.
本文采用密度泛函理论对MTT分子及其银配合物进行结构优化,通过频率计算,绘制拉曼光谱,结合VEDA4软件利用势能函数分布(PED)对拉曼光谱进行了指认和归属[9]. 此外分析讨论了MTT分子的表面静电势和前线轨道,结合TDDFT计算结果分析了Ag3-MTT的吸收光谱、激发态,利用TDDFT的计算数据绘制了电荷转移光谱(Charge-transfer Spectrum,CTS).
2 理论计算
理论计算采用Gaussian09量子化学程序包[10],分子构型用Gauss View6.0构造,对MTT和其银配合物分子的几何结构进行优化,进行了频率计算. 前线轨道和分子表面静电势借助Gauss View6.0程序完成;电荷转移光谱使用多功能波函数Multiwfn软件[11]完成.
3 结果与讨论
3.1 分子结构
根据MTT分子的结构,采用密度泛函理论,B3LYP杂化泛函,H,C,N,S等原子使用6-31g(d)基组,Ag原子使用赝氏基组水平下对MTT、Ag-MTT(S)、Ag-MTT(N4)、Ag-MTT(N5)、Ag-MTT(N6)、Ag-MTT(N7)、Ag-MTT(N8)进行结构优化,优化结果不存在虚频,表明结构是稳定的,S-Ag、N4-Ag、N5-Ag、N8-Ag的键长分别为0.245 nm、0.229 nm、0.217 nm、0.223 nm,其中N5-Ag键长最短即相互作用最强,又因为Ag-MTT(N5)分子的能量比Ag-MTT(S)、Ag-MTT(N4)、Ag-MTT-(N8)分别低1470.33 KJ/mol、17.35 KJ/mol、7.96 KJ/mol,计算结果表明MTT通过N5和Ag能形成更稳定的配合物,图1为优化的MTT及MTT-Ag、MTT-Ag3配合物的结构. 计算结果表明,MTT分子二面角均接近0.00°或180°,表明MTT分子结构为近平面结构. 其中MTT分子C2-N5、N4-N5键长分别为0.133 nm和0.135 nm;Ag-MTT分子C2-N5、N4-N5、Ag13-N5键长分别为0.134 nm、0.138 nm和0.217 nm;Ag3-MTT分子C2-N5、N4-N5、Ag13-N5键长分别为0.134 nm、0.138 nm、0.214 nm. 由上可发现,MTT分子、Ag-MTT分子、Ag3-MTT分子的C2-N5、N4-N5的键长会随着MTT分子连接的Ag数量增多而逐渐变长.
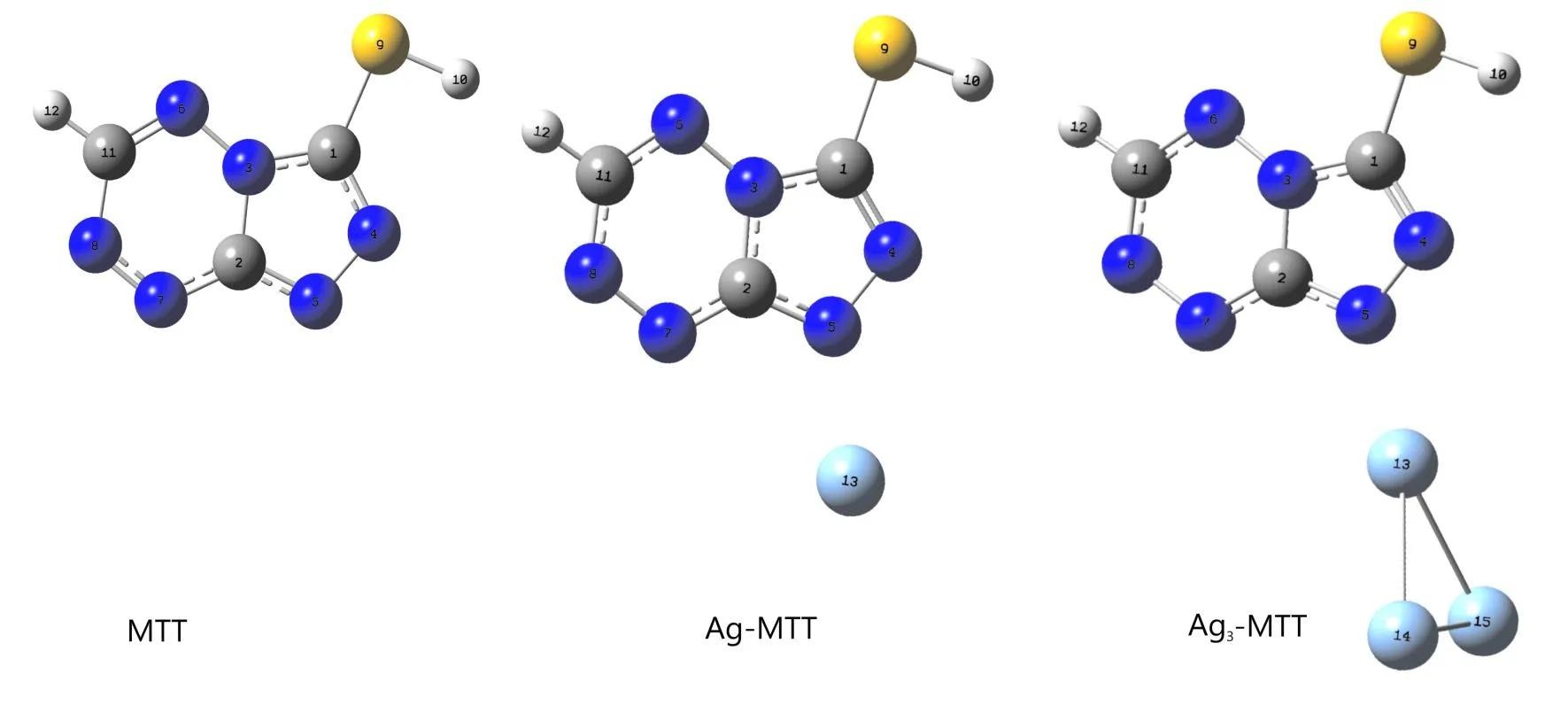
图1 结构优化后的MTT及其银配合物Fig. 1 The optimized structure of MTT,Ag-MTT,Ag3-MTT
3.2 分子振动频率和归属
MTT及其银配物的Raman光谱见图2. 通过势能函数分布结合VEDA4软件,对MTT分子及其银配合物谱图信息丰富的400-1800 cm-1波段进行了指认归属. 1516 cm-1归属于C-N的伸缩振动;1475 cm-1归属于C-N的伸缩振动、四嗪环上H-C-N的面内弯曲振动、C-N-N的弯曲振动;1374 cm-1归属于C-N的伸缩振动、C-N-N的弯曲振动;1327 cm-1、1269 cm-1、1188 cm-1归属于环呼吸振动和面内变形;1119 cm-1归属于C-N伸缩、C-N-N的面内弯曲振动;1060 cm-1归属于N-N伸缩振动、C-N-N的弯曲振动;976 cm-1归属于C-N伸缩、N-N伸缩、C-N-N的面内弯曲振动;946 cm-1归属于C-H的面外弯曲振动;937 cm-1归属于S-H的面内弯曲振动;771 cm-1归属于C-N伸缩、C-N-N的面内弯曲振动;711 cm-1归属于环变形振动;587 cm-1归属于C-N-N面内弯曲振动;449 cm-1归属于C-S的伸缩振动和C-N-N的面内弯曲振动.
分别计算了 MTT、Ag-MTT(N5)、Ag3-MTT(N5)的静态极化率,见表2. 拉曼光谱谱峰的强度和分子极化率对简正振动坐标的微分值大小有关,静态极化率逐渐增大,并且和拉曼光谱谱峰强度的变化趋势也表现出了一致性[12,13].
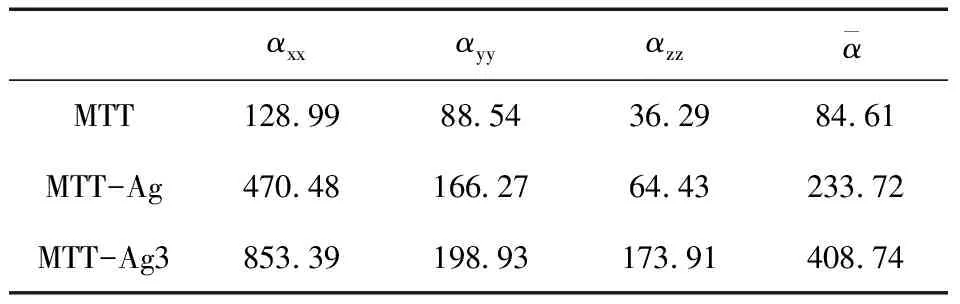
表2 MTT、Ag-MTT(N5)、Ag3-MTT(N5)的静态极化率(a.u.)
3.3 表面静电势
分子静电势分布图(The maping of the molecular electrostatic potential,MEP)对于考察分子间静电相互作用、预测反应位点以及识别分子等方面有重要意义[14]. 红色区域代表负电性集中区域大或亲电区域即具有电负性较大的原子或有孤对电子,蓝色区域代表正电荷集中区域或亲核区域,MTT分子的表面静电势分布图见图3,可以看出MTT分子中的5号N原子上的电荷密度较大,具有极小值-35.62 Kcal/mol. 当MTT和SERS增强基底作用时候,主要通过5号N原子和基底发生作用,与能量和键长判断相符合.

图3 MTT分子的表面静电势Fig.3 The molecular electrostatic potential of MTT
3.4 分子的吸收光谱与激发态
Ag3-MTT的TDDFT激发态计算所得到的理论吸收光谱见图4,有两个明显吸收分布在292 nm和348 nm左右. 通过电荷转移光谱(CTS)的概念[15],旨在讨论从碎片内电荷再分配和片段间电荷转移的角度来理解电子激发的性质. 利用该方法,评估了碎片内电子重分布和片段间电子转移对Ag3-MTT的吸收光谱的贡献,将Ag3-MTT分为Ag3和MTT两个片段,图4中292 nm附近很高的峰主要来自于Ag3片段区域内的电子激发所致,其次体现的是MTT片段向Ag3片段的电子转移效应(ligand-to-metal charge transfer,LMCT)也对这个峰有贡献,Ag3片段也有微弱的向MTT片段的电子转移特性(metal-to-ligand charge trans -fer ,MLCT). 在348 nm附近吸收较为复杂,主要来自于MTT片段向Ag3片段的电子转移和Ag3片段自身的电子激发所致,MTT片段自身的电子激发也有一定贡献. Ag3-MTT为开壳层体系,其结构轨道跃迁、垂直激发能、波长和谐振强度见表3.
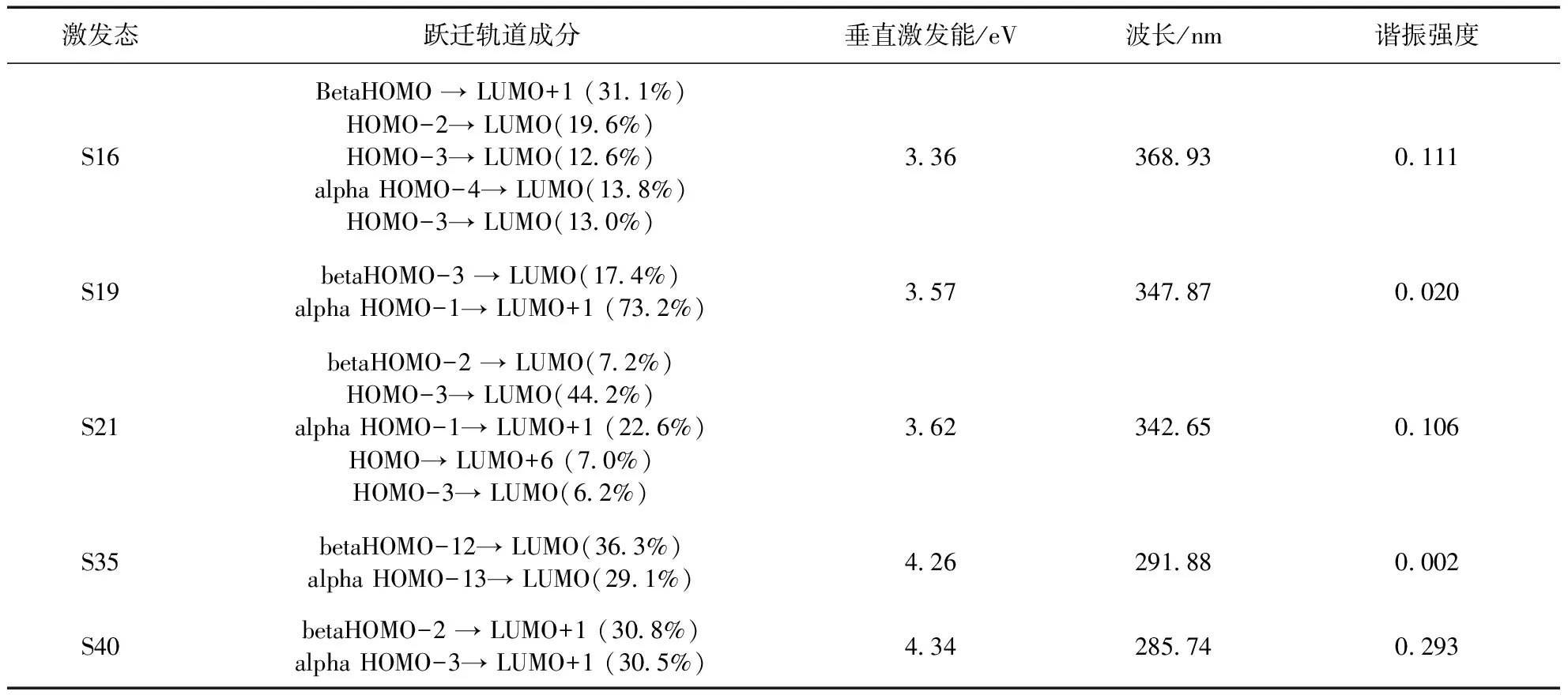
表3 Ag3-MTT的激发态
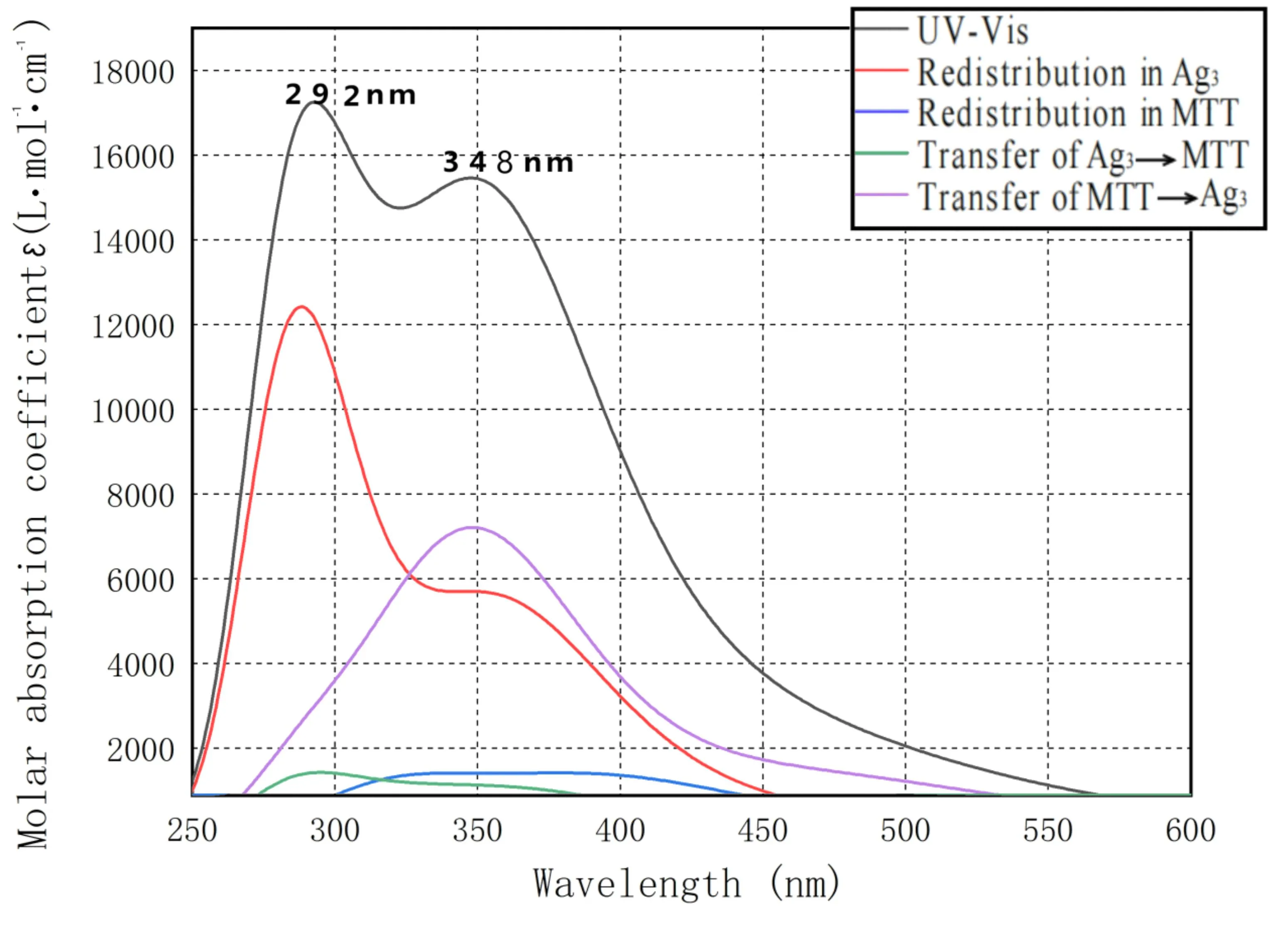
图4 Ag3-MTT的电子吸收光谱和电荷转移光谱(CTS)Fig.4 Electronic absorption spectrum and charge-transfer spectrum (CTS)of Ag3-MTT
292 nm附近的峰主要贡献来源于基态S0跃迁到基态S40(波长为285.74 nm,振幅强度为0.293);348 nm附近的峰的贡献主要来源于基态S0跃迁到基态S16(波长为368.93 nm,振幅强度为0.111)和基态S0跃迁到基态S21(波长为342.65 nm,振幅强度为0.106). 图5为Ag3-MTT的空穴电子分布等值面图,绿色和蓝色分别对应电子和空穴[16],S40的电子和空穴主要集中在Ag3片段上,有小部分电子和空穴在MTT片段上,结果显示292 nm附近的峰主要来源于Ag3片段的局域激发(Local excitation,LE). S16的电子全部在Ag3片段上,而空穴在两个片段上都存在,体现了MTT片段向Ag3片段转移的特性同时也具有Ag3片段内的局域激发的特性;S21的电子主要在Ag3片段上,空穴分布在两个片段上,说明在具有MTT片段向Ag3片段电子转移的特性,与电子转移光谱分析相同.
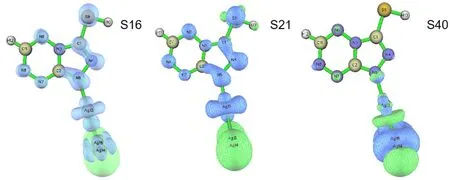
图5 Ag3-MTT的空穴电子分布等值面图Fig.5 The isosurface map of hole electron distribution simultaneously of Ag3-MTT
4 结 论
采用密度泛函理论对MTT分子及其银配合物进行结构优化,讨论了MTT与Ag增强基底之间的吸附方式. 通过频率计算,绘制拉曼光谱,结合VEDA4软件利用势能函数分布(PED)对拉曼光谱进行指认. 分子表面静电势结果表明MTT分子的5号N原子和Ag基底发生相互作用. 并结合TDDFT计算结果分析了Ag3-MTT的吸收光谱和激发态以及电荷转移. 291 nm附近的激发主要来自于Ag3片段自身的电子激发所致,同时Ag3片段向MTT片段的电子转移也有贡献. 346 nm的激发,主要来自于MTT片段向Ag3片段的电子转移和Ag3片段自身的电子激发所致. 本研究结果为研究MTT分子的电子结构、光谱性质和分子识别提供理论依据.
