H2S、HCN、PH3在FeO(100)表面吸附的密度泛函理论研究
2024-02-01彭国建刘永军张艳琨杨春晓夏福婷张秋林
杨 蒙, 彭国建, 刘永军, 张艳琨, 杨春晓, 夏福婷, 张秋林
(1. 云南民族大学 化学与环境学院 云南省高校民族地区资源清洁转化重点实验室, 昆明 650500;2. 昆明理工大学 环境与工程学院, 昆明 650500)
1 引 言
我国是黄磷生产的大国,其生产能力占世界之首,而在黄磷的生产过程中产生大量的黄磷尾气,其含有H2S、HCN、PH3等有毒气体,这些有毒气体的排放不仅污染环境,还严重影响人类的身体健康. H2S和PH3主要作用于人的神经系统,HCN则通过呼吸道影响人类身体健康. 无防护的人群在体积分数为0.05%的HCN下滞留1h时会因身心失调造成危险,当人暴露在浓度为0.03 mg/L的PH3时就会中毒[1-3],因此净化黄磷尾气中的H2S、HCN、PH3是非常必要的. 目前可采用催化氧化法、燃烧法、吸附法等方法净化这三类气体[4-7],三种方法都是利用气体的还原性即通过氧化剂来脱除或者利用其可燃性将其燃烧处理[7]. 催化氧化法作为处理废气的一种有效方法,其具有起燃温度低、操作管理方便等优点,但是其技术流程相对复杂且管理运行成本较高[6,8]. 燃烧法操作较为简单,投入小,但燃烧法一般需要在高温条件下才可以实现对气体的消除,消耗极大的能量,存在二次污染[2]. 而吸附法指通过物理吸附或者化学吸附,将小分子气体吸附固定进而消除[9]. 研究表明[10]通过吸附剂对其进行吸附净化是有效的手段之一,可通过酸或者碱改性炭来脱除黄磷尾气中的H2S和PH3,且经过改性后的吸附体系具有很好的可行性. 活性炭、分子筛、金属及氧化物等对HCN均有较强的吸附作用[6]. 由于使用吸附法净化黄磷尾气具有操作方便、投资小、流程简单的特点[11],所以吸附法在黄磷尾气的净化领域中有非常好的应用前景.
吸附能力越强,催化氧化能力也越强[12],吸附剂在脱除小分子气体中的应用中是一种很有前途的材料. 铁基吸附材料因其价格低廉、吸附活性强和对环境无毒、对于气体分子具有良好的吸附性能等特点而受到广泛关注. 研究表明[13]Fe2O3能有效地控制脱硝过程中HCN的生成,但水蒸气和温度对Fe2O3的催化作用有一定的影响. 田森林等人[14]发现铁基高温变化催化剂与黄磷尾气中的有毒气体发生反应,生成不同的物质从而导致催化剂中毒. 在去除这些小分子气体时由于它们的剧毒性、腐蚀性、实验条件苛刻等特点限制了研究人员对其进行深入广泛的研究. 基于密度泛函理论计算不同反应过程中的吸附能、结构参数,对于理解小分子气体在金属氧化物催化剂上的吸附和解离有着一定的贡献,研究效率大大提高[15,16]. Lin等[17]研究了H2S在α-Fe2O3(001)面的分子吸附和解离吸附过程,发现α-Fe2O3促进了H2S分解成硫物种,这为后续脱硫工艺提供了一定的理论指导. Ag掺杂有利于H2S在Fe2O3表面的解离和吸附,同时结合能也增加,从而提高Fe2O3对H2S的气敏性能[18]. Ren等[19]通过计算表明当HCN吸附在Fe(100)面上的不同的的吸附位点,吸附在Fe(100)面上的Fourfould吸附位点最稳定. 当Fe2O3处于还原气氛时会生成FeO,铁基吸附剂在不同的价态中表现出不同的吸附特性. 陈曙[20]等人研究发现气相FeO分子催化乙炔三聚环化合成苯的反应机理,该反应在热力学上是有利的. Robina Ashraf 课题组利用密度泛函理论研究了FeO的结构和电子性质,通过计算发现与LDA+U相比,GGA+U给出了更准确的结果[21]. FeO与国内外的其他金属催化剂相比,也具有原料丰富,价格低廉的优点[22]. 前人研究了H2S和HCN在其他铁基吸附剂上的吸附性质,但H2S、HCN和PH3在FeO上的吸附理论研究却未见报道. 所以本文基于密度泛函理论,研究黄磷尾气中H2S、HCN、PH3在FeO(100)表面的吸附特性,通过考虑所有可能的吸附位点,并计算分析各分子的吸附能、结构参数、电荷转移、态密度,找出H2S、HCN、PH3在FeO(100)表面上的最佳吸附位点,进一步为净化黄磷尾气的吸附剂提供一定的理论指导.
2 计算方法及模型
研究表明FeO(100)表面的结合能最低[23,24],该表面是最稳定的,所以本文中所有的计算都在FeO(100)面上进行. 本文所有的理论计算都采用Materails-Studio软件包中的Dmol3软件包完成,在Dmol3中,电子交换关联势使用广义梯度近似法(GGA)的PBE来描述,选取FeO(100)表面作为表面模型,构建2×2晶胞[25],为避免各平板间的相互作用,真空层设置为12 Å,固定底部两层原子,使其余三层原子处于驰豫状态,K点设置为4×4×1,每个原子之间的受力最大是0.004 Ha/Å,最大位移为0.005 Å,在所有的计算中smearing参数为0.008Ha,DNP(基组截断)为3.5,计算精度为良好,原子核内电子采用全电子处理. 吸附能计算公式表示为Eads=Egas+FeO-Egas-EFeO[26],其中Eads为吸附能,即吸附前后能量的变化,Egas-FeO为气体吸附在FeO(100)表面时体系的总能量,Egas为气体优化后的能量,EFeO为FeO(100)表面优化后的能量. 吸附能可用于判定吸附后吸附体系的稳定性:吸附能越负,该吸附体系越稳定.
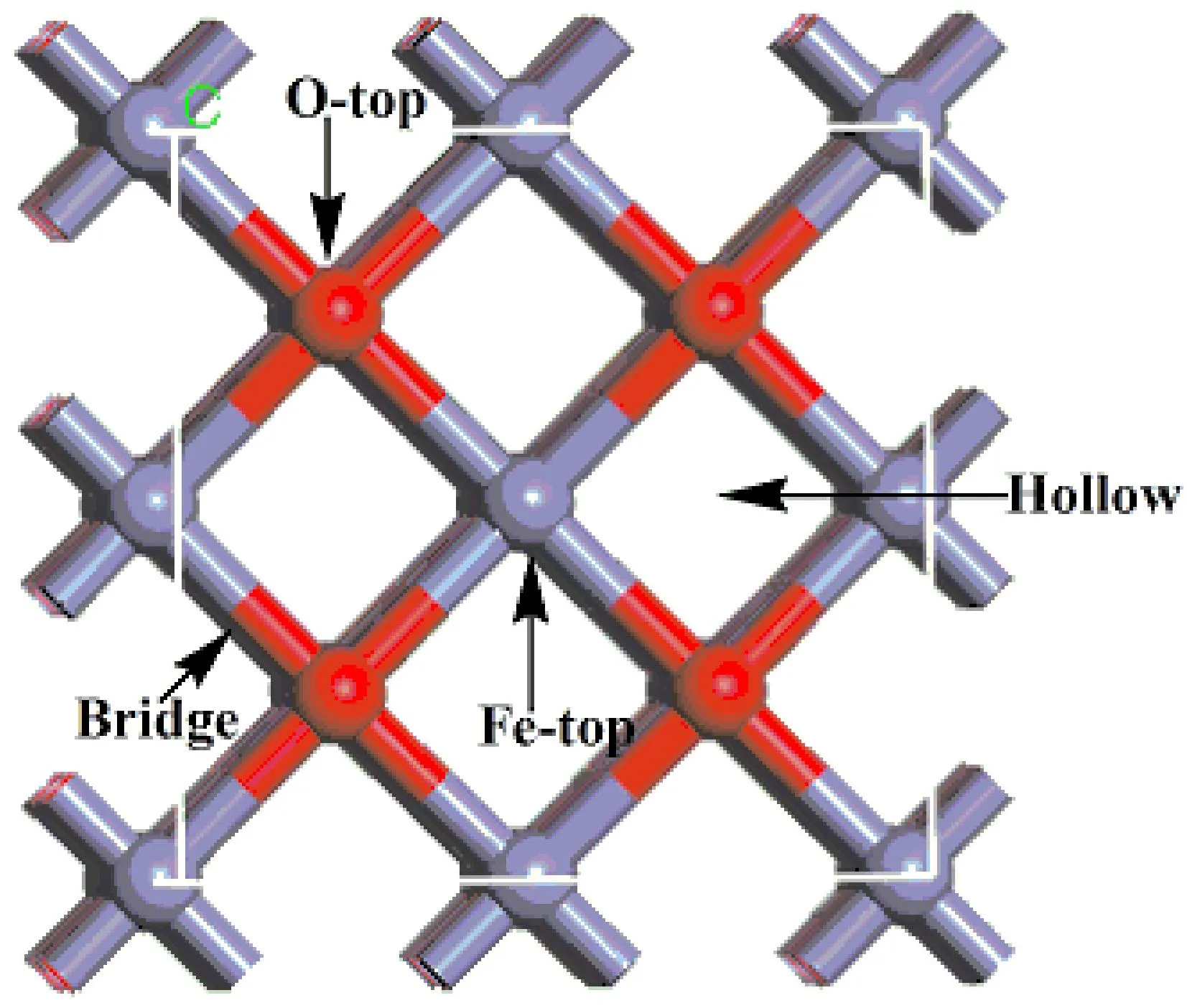
图1 FeO(100)面吸附位点的俯视图Fig. 1 Top view of the adsorption site on theFeO(100)surface
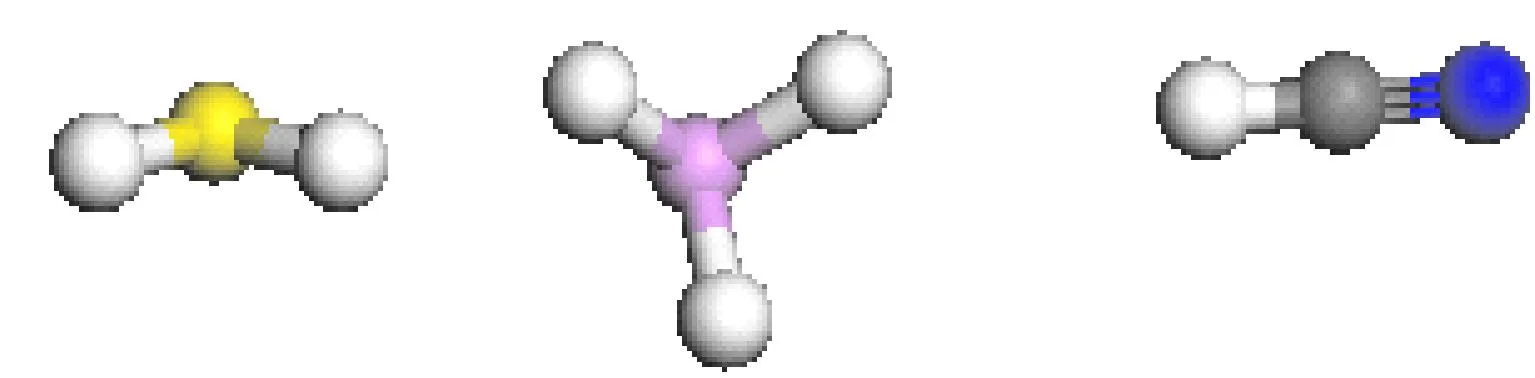
图2 优化后的H2S、PH3、HCN分子Fig. 2 The optimized H2S,PH3,HCN molecules
3 结果与讨论
3.1 H2S在FeO(100)表面的吸附
3.1.1H2S在FeO(100)表面的几何结构和吸附能
对于气相H2S分子,通过计算所得的H-S键为1.357 Å,∠HSH为92.2°,与实验值[15]1.328 Å和91.8°较为吻合,所以计算方法是合理的. Wang等人[27]研究表明硫原子吸附在金属氧化物表面时有很好的吸附强度,所以本文中H2S所有的吸附模型都是硫原子以垂直方式吸附在FeO(100)面上的各吸附位点,表1给出了H2S在FeO(100)表面上各吸附位点的参数信息. 通过比较表1中的吸附能可得到H2S在FeO(100)表面各吸附位点的稳定顺序如下:O-top>Hollow>Fe-top>Bridge,O-top吸附位点为化学吸附,Hollow吸附位点是弱化学吸附,且两者的吸附能相差不大,而其它位点的吸附为物理吸附. 吸附在O-top的H2S中H-S键与未吸附之前相比增加了0.005 Å,且在该吸附位点的H-S-H的角度有所增加,说明H2S中H-S键在该吸附位点上被活化,可能导致H-S键的断裂[14],而其它吸附位点的H2S中H-S键与自由分子H2S的键长相比变化不大.
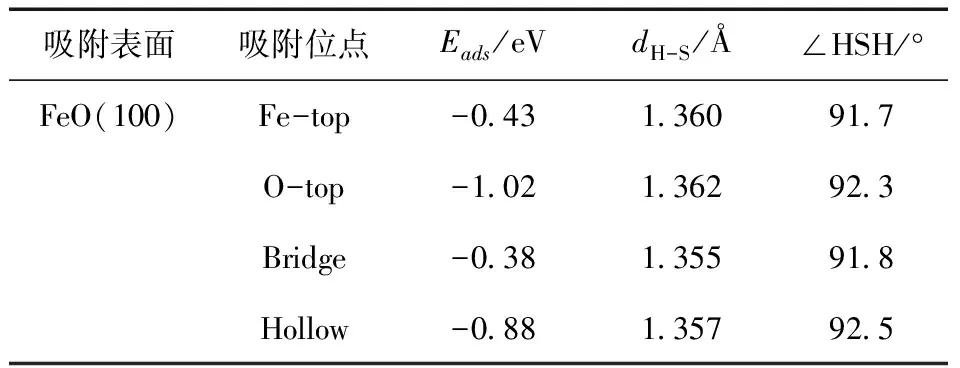
表1 H2S在FeO(100)表面的吸附能和吸附相关参数
3.1.2Mulliken电荷布居分析
表2列出了理想H2S气体的Mulliken电荷分布和吸附于FeO(100)不同吸附位点的的Mulliken电荷分布. 表2列出了吸附后H2S中各原子的电荷变化量以及电荷变化量的总和,QH2S为负值表示获得电子,为正值意味着失去电子[28]. 宋佳佳[29]等研究表明:吸附后转移的电荷量越少表明吸附越弱. 通过分析可得吸附在O-top吸附位点后H2S的Mulliken电荷的总和(QH2S)变为0.126 e,是四个吸附位点中转移的电荷量最多的一个吸附位点,同时也表示电荷从H2S转移到FeO(100)面,增强了其表面的吸附能力,说明H2S在O-top吸附位点的吸附是最强的,也即是最稳定的. 当H2S吸附在FeO(100)表面上的四个吸附位点时,Bridge吸附位点和Hollow吸附位点的H、S原子的电荷变化量都为正值,而Fe-top吸附位点和O-top吸附位点的S原子的电荷变化量是负的,表示H2S气体分子中S的电荷转移到FeO(100)表面上,增强了表面的吸附能力,且O-top的S原子的电荷变化量是最多的,说明该位点的吸附能力最强,这与吸附能分析的结果一致.
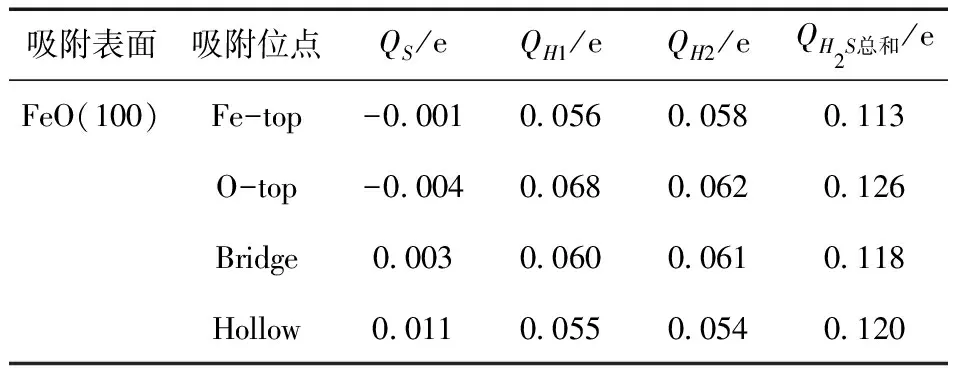
表2 H2S吸附在FeO(100)表面的电荷转移
3.1.3态密度分析
为了进一步分析吸附机理,本文将利用Dmol3中的Analysis功能来分析H2S吸附在O-top吸附位点的态密度. 如图3所示:A图为H2S吸附前后的态密度,从A图可得到吸附后的H2S态密度向低能级移动,说明吸附体系更稳定[16]. 从B图中可知H2S的S原子的3p轨道吸附后的态密度向更负的能级移动,且峰值较吸附前变弱,表明H2S与FeO(100)表面的成键作用强[30]. 吸附后的S原子的p轨道与FeO中Fe原子的d轨道在能量为-2.5 eV处出现了一个杂化峰,说明FeO(100)表面的该吸附位点与H2S的相互作用较强.

图3 H2S吸附在O-top吸附位点的态密度Fig. 3 Density of states of H2S adsorbed on O-top adsorption site
3.2 HCN在FeO(100)表面的吸附
3.2.1HCN在FeO(100)表面的几何结构和吸附能
通过计算得到HCN的C-H键键长为1.079 Å,C-N键的键长为1.162 Å,HCN键角为179.3°,与实验值1.064 Å、1.156 Å、180°较为接近[31],说明计算方法是合理的. HCN易于通过具有孤对电子的N原子垂直吸附在金属表面上[32],所以本文只考虑N原子垂直吸附. 表3给出了HCN在FeO(100)表面上不同吸附位点的吸附能和吸附相关参数,通过吸附能的比较可得出其稳定顺序为Hollow>Bridge>Fe-top >O-top. HCN在O-top吸附位点上的吸附属于物理吸附,吸附后的C-N键与吸附之前的相比,没有发生任何变化,H-C-N键角变化也不大,说明该吸附位点上HCN的吸附很不稳定. 当HCN吸附在Hollow位点时,属于弱化学吸附,C-N键由原来的1.162 Å变为1.165 Å,被拉长了0.003 Å,C-N键的拉长表示该位点具有催化活性,H-C-N键角由原始的179.3°变为179.7°,键角变化率为0.22%,前人研究表明:HCN的键角变化率越大,表面对HCN的吸附能力越强[32]. 所以Hollow位点为HCN的最稳定吸附位点.
3.2.2Mulliken电荷布居分析
采用Mulliken电荷分析方法研究了FeO(100)表面与HCN气体之间的电荷转移情况,进一步说明FeO与HCN之间的相互作用机理,表4给出了HCN吸附在FeO(100)表面上的不同吸附位点的电荷转移. 从表中可以看出:Hollow吸附位点的电荷变化量是最大的,其次是Bridge吸附位点,变化最小的是O-top吸附位点,电荷变化量越大,吸附作用越强,因此Hollow吸附位点的吸附能力是最强的,而O-top吸附位点的吸附能力是最弱的,这与吸附能分析结果相对应.
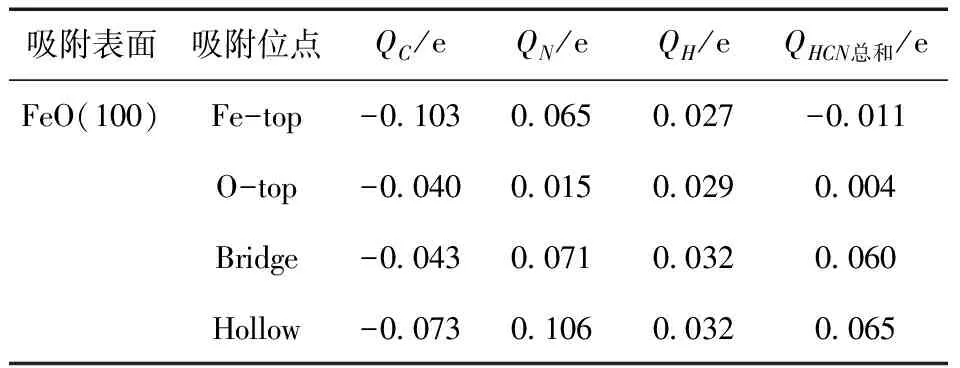
表4 HCN吸附在FeO(100)表面的电荷转移
3.2.3态密度分析
图4中的A图为HCN吸附在Hollow位点上的态密度,由图可得:吸附后的HCN的态密度向低能级移动,说明吸附后的HCN比气相中的HCN更稳定. 从B图可看出:吸附后的p轨道向低能级移动. 而C图中吸附后的d轨道在费米能级处的峰值明显下降,表明在吸附过程中,HCN中N的p轨道与FeO中Fe的d轨道形成N-Fe键.
B图中吸附后N原子的p轨道与C图中吸附后Fe原子的d轨道在能量-8.2 eV处有很小的杂化峰,表示S原子与FeO(100)表面的最外层的Fe原子发生了相互作用.
3.3 PH3在FeO(100)表面的吸附
3.3.1PH3在FeO(100)表面的几何结构和吸附能
有研究表明[32]当P原子垂直吸附时,其结合能最低,更有利于吸附. 所以本文只考虑P原子的垂直吸附. 表5总结了PH3在FeO(100)表面的吸附特点,通过对比吸附能可得:PH3在FeO(100)表面最稳定的吸附位点为O-top,吸附能为-1.11 eV,PH3在Hollow吸附位点的吸附能与O-top吸附位点相近,其吸附能为-1.10 eV. PH3在FeO(100)表面最不稳定的吸附位点是Fe-top,属于物理吸附. PH3的P-H在O-top吸附位点上的键长较未吸附之前有一定的变化,且键角变化是最大的,表明该吸附位点有很强的吸附能力.
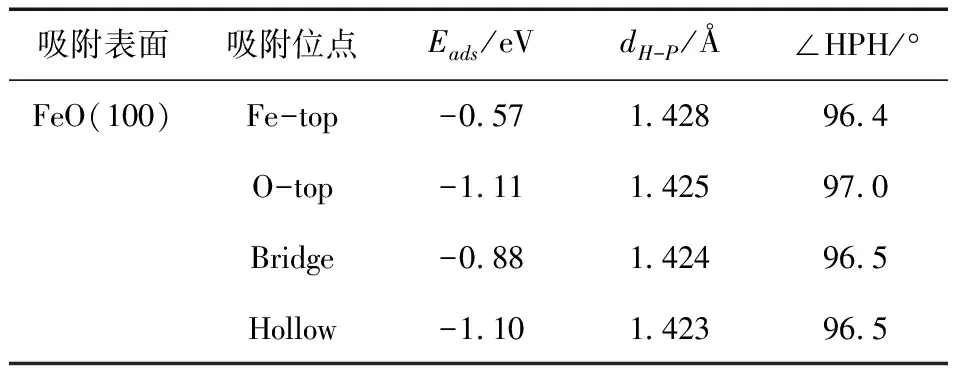
表5 PH3在FeO(100)表面的吸附能和吸附相关参数
3.3.2Mulliken电荷布居分析
从电荷转移变化量来证实前面通过比较吸附能大小所得到的结果. 从表6中可得到:吸附在O-top吸附位点上的电荷变化量是最多的,表明PH3与FeO(100)表面间的成键作用强,也即吸附最稳定的,而Hollow位点的电荷变化量与O-top吸附位点的较为接近,稳定性也较为稳定,这与吸附能的结果一致.
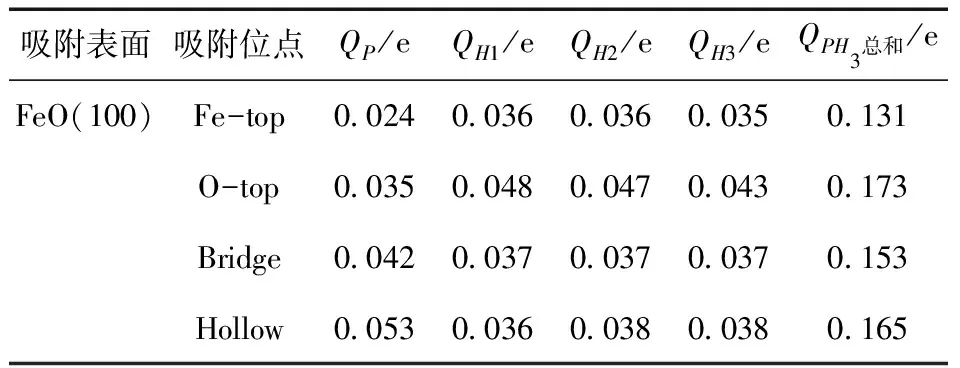
表6 PH3吸附在FeO(100)表面的电荷转移
3.3.3态密度分析
图5为PH3吸附在O-top吸附位点的态密度,利用态密度可以进一步理解PH3在FeO(100)表面的吸附作用. 由A图可知:吸附前的态密度主要分布在-9.2 eV、-2.6 eV、0 eV处,而吸附后的态密度主要在-13 eV、-6.6 eV、-5.5 eV、-5.4 eV、-1.2 eV,吸附后PH3的态密度整体向更低能量区域偏移大约3.8 eV,且态密度峰值降低,说明吸附后的PH3更稳定[34]. 吸附后PH3中P原子的p轨道与FeO中Fe原子的d轨道的峰值下降,说明PH3与FeO(100)表面之间的结合能力增强,吸附体系更稳定[35,36].
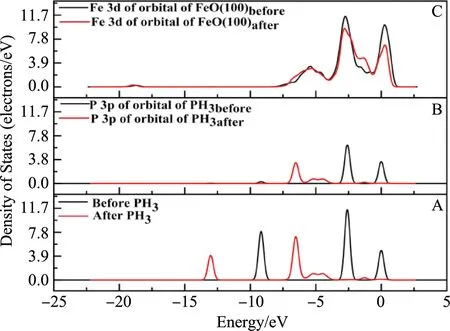
图5 PH3吸附在O-top吸附位点的态密度Fig. 5 Density of states of PH3 adsorbed on O-top adsorption site
4 结 论
本文采用密度泛函理论研究了H2S、HCN、PH3在FeO(100)面的吸附,通过分析三种小气体分子在FeO(100)表面上不同吸附位点的吸附能、电荷转移量和最稳定的吸附位点的态密度,得到以下结论:当H2S、PH3分别吸附在FeO(100)面上的四个吸附位点时,O-top吸附位点的吸附能最负,即该位点是H2S和PH3的最稳定吸附位点,且H2S吸附在O-top吸附位点时,H-S键有所伸长,说明H2S在FeO(100)表面上发生了H-S键的伸长,而HCN最稳定的吸附位点为Hollow位点,吸附能为-0.8 eV. H2S、HCN、PH3在最稳定吸附位点时所转移的电荷量是最多的,说明该位点的吸附结构较稳定. HCN在Hollow吸附位点和PH3在O-top吸附位点时的键角较未吸附之前的变化是最大的,表示该吸附位点有较强的吸附能力;H2S、HCN、PH3分别吸附在FeO(100)表面的最稳定吸附位点时,态密度与吸附前的态密度相比,都往更低的能量移动,且吸附后的态密度的峰值有所降低,吸附体系变得更加稳定,与吸附能分析的结果一致.
