先天性外胚层发育不良一家系基因突变检测
2023-10-24葛新红马迎东刘玲玲
紫 薇 葛新红 马迎东 刘玲玲 周 镁
1宁夏医科大学,宁夏银川,750003;2宁夏医科大学总医院,宁夏银川,750003
临床资料先证者,男,49岁。因“全身毛发稀疏、无汗液分泌伴部分牙齿缺如49年”就诊。 现病史:先证者自诉在出生时全身水肿、无毛发,易哭闹,出生2天后水肿逐渐消退,随后全身皮肤反复脱屑,伴间断性发热(体温不详),伴腹泻、呕吐,曾多次就诊当地医院及卫生所,诊断不详,先后予以多种抗生素及中药治疗无明显疗效,于1岁时上述症状逐渐缓解。约4岁时开始出牙(共8颗牙齿,至今未更换过牙齿),头皮逐渐有新生稀疏毛发长出。先证者长期怕热,无汗液分泌,皮肤干燥,双眼干涩,环境温度升高时自觉症状加重。于2022年2月11日就诊于我科门诊,考虑“先天性外胚层发育不良”,进一步完善皮肤组织病理学和基因检测。 既往史:先证者10年前曾患“肺部感染、肾病综合征、脂肪肝、胆囊壁增厚”,现肺部感染已治愈。 家族史:先证者母亲有类似病史、风湿性心脏病、三尖瓣和主动脉瓣膜置换术后;先证者女儿部分牙齿缺失,智力评级为智力二级残疾(图1)。先证者父母非近亲结婚。
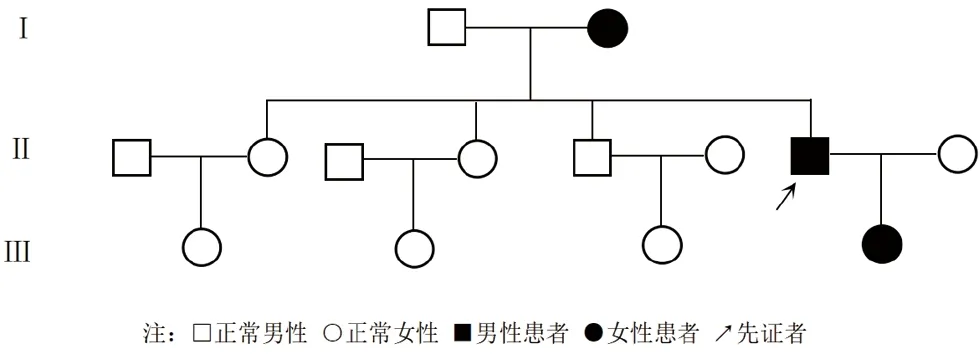
图1 先证者家系图
皮肤科检查:头发稀疏,头顶部、两侧颞部头发缺如,眉毛、腋毛和外阴部毛发稀少;鼻梁凹陷;上牙槽可见四颗尖牙,三颗义齿;下牙槽可见三颗磨牙,部分牙齿呈黑褐色或锥形;躯干及四肢未见毳毛,双手指甲甲床发白(图2)。先证者女儿牙齿发育不良,形状为锥形,部分牙齿缺失(图3)。

2a~2e:鼻梁塌陷,眉毛缺如,头顶部、两侧颞部及枕部头发稀少;2f~2h:牙齿稀疏,上牙槽可见四颗尖牙,三颗义齿,下牙槽可见三颗磨牙,部分牙齿表面呈黑褐色;2i、2j:躯干、双上肢皮肤未见毳毛;2k:双手指甲甲床发白
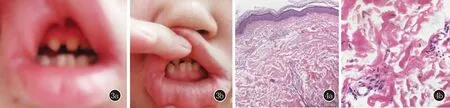
图3 先证者女儿牙齿表现(患者提供) 3a、3b:患者女儿牙齿发育不良,形状为锥形,部分牙齿缺失 图4 先证者右上肢皮肤组织病理 4a:表皮角化过度,未见皮肤附属器(HE,×100);4b:真皮层血管周围少量淋巴细胞浸润(HE,×400)
皮肤组织病理学表现:先证者(右上肢)皮肤组织,表皮网篮状角化,真皮层血管周围少量淋巴细胞浸润,未见皮肤附属器(图4)。
基因检测结果及分析:通过对疾病相关基因的测序分析,发现先证者突变位点为EDA c.463C>T(p.R155C),即EDA基因有1个半合子突变,在463号核苷酸由胞嘧啶C变为胸腺嘧啶T(c.463C>T)的半合子突变,导致第155号氨基酸由精氨酸变为半胱氨酸(p.R155C),为错义突变。该变异对应的表型是X连锁少汗性外胚层发育不良、X连锁选择性牙齿发育不全1型,根据美国医学遗传学与基因组学会(The American College of Medical Genetics and Genomics, ACMG)发布的变异解读指南,该变异初步判定为疑似致病性变异。经Sanger验证,先证者母亲及女儿该位点杂合变异,先证者父亲该位点无变异。先证者另一可疑变异位点PRKD1 c.535+3A>G(splicing),经查询该位点位于内含子位置,该变异对应的表型是先天性心脏缺陷和外胚层发育不良,根据ACMG指南,该变异初步判定为临床意义未明。经Sanger验证分析,先证者母亲的该位点为杂合变异,先证者父亲及女儿该位点无变异。而以上变异的来源是先证者母亲(图5)。
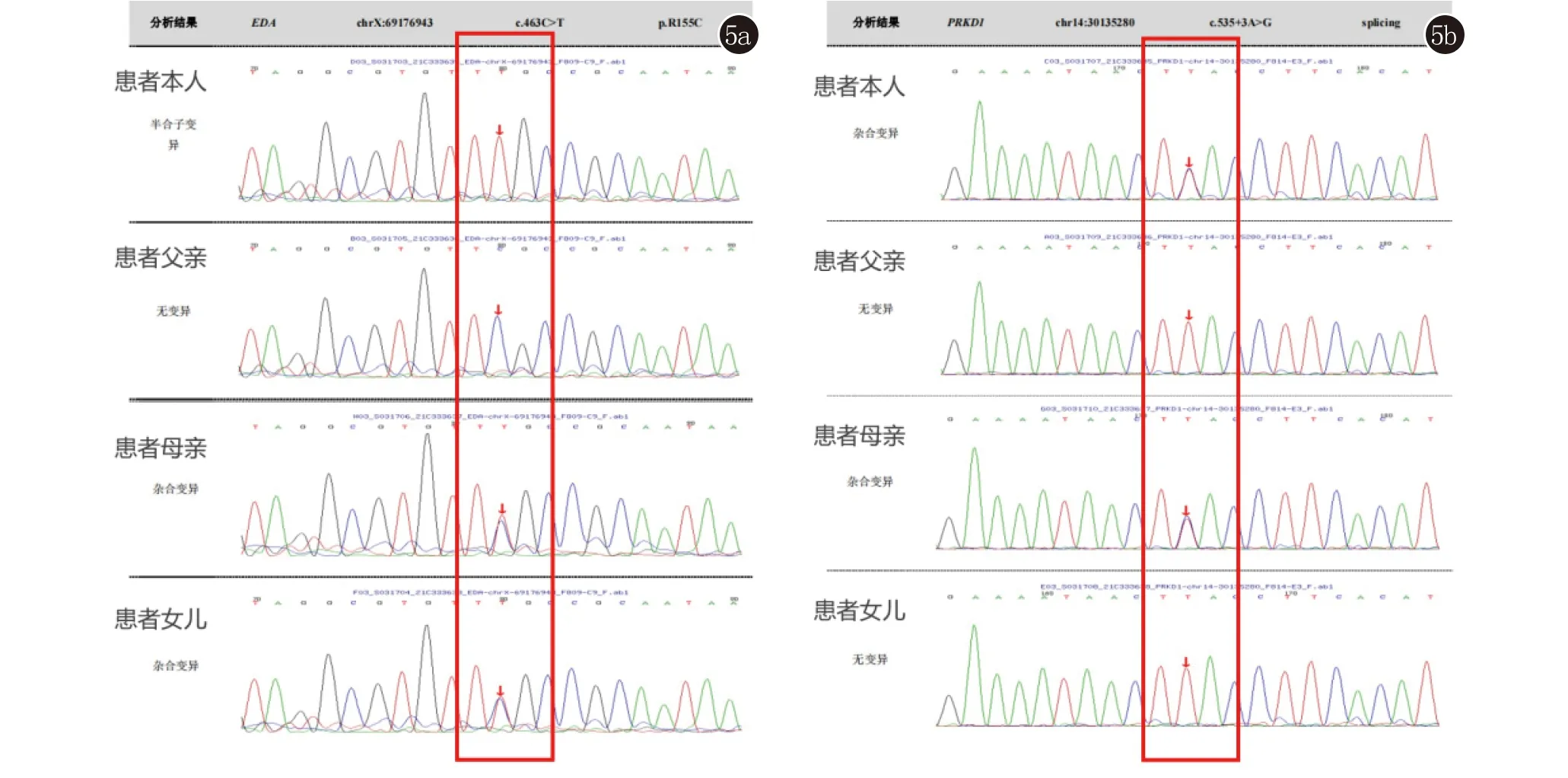
5a:致病突变位点EDA c.463C>T(p.R155C);5b:可疑突变位点PRKD1 c.535+3A>G(splicing)
最后诊断:先天性外胚层发育不良。
讨论外胚层发育不良(ectodermal dysplasia,ED)是指一组罕见的异质先天性疾病,影响外胚层结构的正常发育,包括皮肤、头发、牙齿、指甲和小汗腺等[1]。临床上通常根据小汗腺发育是否正常将ED分为少汗型(无汗型)ED和有汗型ED[2]。少汗型外胚层发育不良(hypohidrotic etcodermal dysplasia,HED)是ED最常见的形式[3],大多为X连锁遗传,是由X染色体上外异蛋白A(ectodysplasin A,EDA)基因的致病变异引起的,EDA信号通路在胚胎外胚层发育中起重要作用[4],EDA基因突变导致外异蛋白A1(ectodysplin A1,EDA1)的缺失或功能障碍,从而形成典型的少毛症、缺牙和少汗或无汗三联症[5]。
在一项为期5年的对X-连锁的少汗性ED(XLHED)患者的自然史研究[6]证实,XLHED的女性患者与男性患者相比症状更轻,但女性患者均表现相关的症状。在此病例中,先证者的症状比其母亲的症状更加明显,先证者女儿存在牙齿缺失及牙齿锥形改变,智力亦受到影响。其他与XLHED相关的问题,如乳房发育不足及其影响(母乳喂养困难和社会心理问题)在EDA突变的女性携带者中也很常见[7],我们在询问先证者病史过程中了解到其母亲也曾存在母乳喂养困难。
对这种罕见疾病的产前诊断可为终止妊娠提供基础[8]。产前诊断要结合病史、家族史、非侵入性超声检查结果和产前基因检测[5]。李亮等[9]报道了一例29岁有ED家族史的女性患者,在其第三、四次怀孕时接受了胎儿基因检测及牙胚超声检查,发现胎儿存在EDA基因突变及牙胚结构的异常,最终选择终止妊娠。而产前诊断为产前治疗提供了可能[8]。在Margolis等[10]的研究中,将重组外胚层发育不良蛋白(Fc:EDA1)注入犬模型受影响胎儿的羊水中,并在犬胎儿牙胚及毛囊等发育的关键时期进行多次重组蛋白的治疗,可明显改善新生犬的症状。在Schneider等[11]的一项报道中,他们在孕妇妊娠中晚期将Fc:EDA1通过羊膜腔给药治疗受影响的胎儿,发现第33周(双胞胎)和第39周(单胎)婴儿出生后能够正常出汗,并且在婴儿14~22月龄时与XLHED相关疾病并未发展。虽然羊膜穿刺术过程中会有一定概率导致孕母体和胎儿因手术创伤或医源性细菌感染而流产[12],但笔者认为产前治疗仍为此类患者提供生育健康后代的可能性。
与先证者的交谈过程中,我们了解到该疾病对患者的工作及生活产生巨大的影响。患者因丧失排汗功能而无法耐受高强度的工作,以及牙胚发育不良无法进行义齿的完全矫正,从而影响了咀嚼等基本的口腔功能,且因患者腺体发育缺陷,肺部感染的几率增加。此前先证者对该疾病并不了解,对女儿智力发育的影响让其抱憾终生。我们建议先证者在生活中尽量避免高温环境,采用各种散热措施,并避免感染发生。另外,我们已经明确先证者EDA基因有一个半合子突变EDA c.463C>T(p.R155C),鉴于该疾病的遗传特点,建议先证者的亲属如有生育后代的需要,可先行基因检测或产前诊断,以提高孕育健康后代的概率。
