一个两次多囊肾胎儿孕育史家系的临床分析及遗传咨询
2019-06-03吴庆华苏国玲麻希洋梅世月孔祥东史惠蓉
吴庆华,王 参,苏国玲,麻希洋,梅世月,孔祥东,史惠蓉
郑州大学第一附属医院遗传与产前诊断中心 郑州 450052
多囊肾病(polycystic kidney disease,PKD)是一种临床表型和遗传异质性较高的疾病,分为常染色体显性遗传PKD(autosomal dominant PKD,ADPKD)和常染色体隐性遗传PKD(autosomal recessive PKD,ARPKD)。最初ADPKD被认为只在成人期发病,故又被称为成人型PKD,而绝大多数ARPKD只在婴儿期发病,故临床上也称为婴儿型PKD,目前有报道[1-2]表明ADPKD也可在儿童期甚至婴儿期发病,而ARPKD可在出生后或更晚时期发病。ADPKD发病率估计为1/1 000~1/400[3],半数患者在70岁前进展为终末期肾衰竭,85%的ADPKD病例是由于16p13.3~13.12上的PKD1基因突变致病,15%为4q21~23上的PKD2基因突变引起。ARPKD在活产儿发病率约为1/20 000,位于6号染色体p12.3~12.2的PKHD1是其主要致病基因[4]。因患者临床症状及发病时间变异大,PKD的确诊需明确基因诊断,大多数患者经高通量测序技术而被诊断。本研究家系中,先证者妻子曾有两次多囊肾胎儿孕育史。为避免先证者家庭再次孕育患儿,我们应用高通量测序技术对先证者及其妻子行遗传性肾病相关基因筛查,对该家系相关成员行基因分析,以明确病因,为再次妊娠提供指导。
1 对象与方法
1.1研究对象先证者及其配偶妊娠两次,均于孕中期因胎儿超声提示多囊肾,羊水过少而终止妊娠且未留取胎儿组织标本,再次妊娠前来我院行遗传咨询了解生育风险。该夫妇无近亲结婚史、无明确的孕期药物或不良因素接触史。本研究获我院伦理委员会批准(批准号:KS-2018-KY-36),研究对象均知情同意。
1.2家系调查先证者,男,26岁,无特殊临床表现,在我院经彩超及CT检查提示双侧肾脏大小形态正常伴多发囊肿,尿常规提示尿蛋白1+。先证者妻子腹部彩超、尿常规及生化检查均未见异常。先证者母亲,53岁,彩超检查提示双肾大小形态正常伴多发囊肿,无与多囊肾相关的临床表现及其他不适。先证者父亲腹部超声未见异常。先证者姐姐,29岁,彩超提示双肾形态欠规整,右肾可见一大小约7 mm×5 mm的囊肿。先证者弟弟,23岁,于2 a前曾因“腹痛,腹部包块”来我院就诊,血压高至159/101 mmHg(1 mmHg=0.133 kPa),超声提示双肾形态失常并多发囊肿;泌尿系计算机体层摄影尿路造影检查发现肾形态失常,双肾多发囊肿,左肾为著,双肾多发结石;肾动态显像和肾小球滤过率检查提示肾小球滤过率下降,左右侧分别为33.58及49.48 mL/min;尿常规示白细胞3+,蛋白2+;肾功能示尿素9.88 mmol/L,肌酐133 μmol/L;心脏超声提示左室壁均匀性增厚(高血压性心脏病样改变)。先证者岳父母肝肾超声未见异常。该家系遗传图谱见图1。
1.3遗传性肾病基因筛查
1.3.1 DNA提取 采集该家庭先证者及其配偶,先证者母亲、姐姐及弟弟的外周血2 mL,采用北京天根公司的DNA提取试剂盒提取全基因组DNA,4 ℃保存。
1.3.2 高通量测序 采用靶向基因捕获技术结合Illumina二代测序平台,对先证者及其配偶行63个遗传性肾病相关致病基因(肾脏和泌尿系发育异常、肾小球疾病、肾小管疾病、纤毛功能异常、遗传代谢病相关的基因)的高通量测序。
1.3.3 PCR及Sanger测序 高通量测序分析发现PKD1基因为可疑致病突变位点,应用PCR扩增先证者及其母亲、姐姐及弟弟全基因组DNA,并用Sanger测序进行一代验证,应用Primer 5软件设计引物,上游引物序列:5’-CAAGAGGCTCAAGAAACTGCCCG-3’,下游引物序列: 5’-GGCCACCAGT GAGAAGTACAGG-3’,引物由生工生物工程(上海)股份有限公司合成。采用ABIBigDye 3.1测序试剂盒对产物进行双向测序,用ABI Sequencing Analysis 5.1.1软件将测序结果与正常序列进行比对,验证基因突变位点。
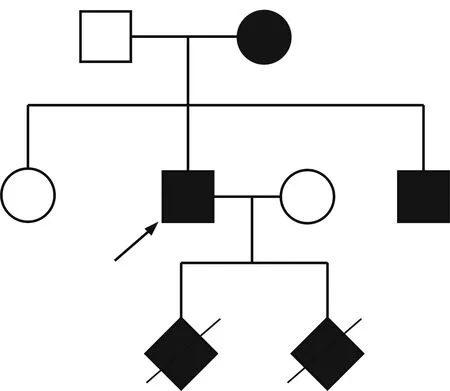
□:正常男性;■:患病男性;○:正常女性;●:患病女性;:先证者;:终止妊娠的患胎
图1该家系遗传图谱
2 结果
2.1高通量测序结果先证者的PKHD1基因各外显子序列可检测到多个已知多态性变异,未检测到已知或者疑似致病变异;其PKD1基因检测到1个疑似致病突变:c.10678G>A(p.G3560R)杂合突变,同时检测到多个多态性变异,未见其他明确与该疾病相关的基因突变;先证者配偶未检测到可疑突变。
2.2PCR和Sanger测序结果对先证者及其母亲、姐姐及弟弟行PKD1基因c.10678G>A突变筛查,结果确定先证者及其母亲和姐姐携带PKD1基因c.10678G>A杂合突变;先证者弟弟未检测到该突变,见图2。
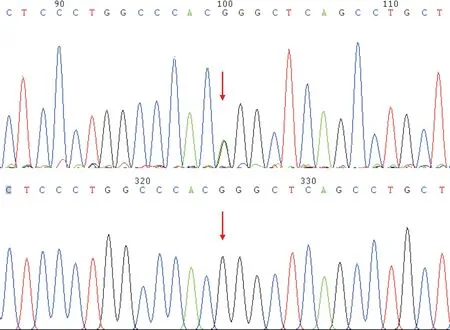
上:箭头示先证者PKD1基因c.10678G>A(p.G3560R)杂合突变;下:先证者弟弟该位点未见突变
图2先证者及其弟PKD1基因c.10678G>A的突变筛查结果
3 讨论
绝大多数ARPKD患者在胎儿期即被诊断,常表现为双侧显著增大的肾脏及羊水过少,由于肾脏体积过大压迫胸腹腔而导致患儿肺脏发育异常,高达30%~50%的患儿在出生后不久即死于呼吸道功能不全,而肾功能衰竭很少是新生儿的死亡原因[5]。存活者有肾脏功能受损且通常伴肾性高血压和门静脉高压,可导致新生儿期及儿童期死亡。PKHD1基因突变是导致ARPKD的主要病因,有研究表明DZIPIL可能是另一个与其发病相关的基因[6-8]。
ADPKD多在出生后人群中表现为肾脏囊肿,增大的肾脏体积与肾功能受损程度呈正相关[9-10],患者肾脏功能受损表现为尿浓缩功能下降,随后出现肾功能不全等,还可有因感染或囊内出血导致的急性或慢性疼痛、高血压、血尿、尿路感染、肾结石等以及肾脏外表现如肝囊肿等[11-12]。患者疾病进程变异大,绝大多数患者起病于20~40岁,仅2%~5%在儿童期确诊[6],少数患者在胎儿期发病,已有相关文献[13]报道早发型ADPKD(儿童期甚至胎儿期发病)主要由PKD1基因突变所致。ADPKD具有高度临床异质性,即使同一家庭中携带相同基因突变者临床表现也可以存在很大差别[14]。尽管ARPKD不如ADPKD常见,遗传及临床表型异质性不如ADPKD高,但有少数ARPKD患者可存活至儿童期甚至成人期,极少数患者可存活至老年才发病,故少数情况下ADPKD较难与ARPKD相鉴别[15-18]。
本研究家系中,先证者双肾多发囊肿,该夫妇有两次多囊肾胎儿孕育史且均未留取组织标本,来我院进行产前遗传咨询。我们首先应用高通量测序技术对先证者及其妻子行包括PKD在内的63种遗传性肾病基因筛查,结果提示先证者PKD1基因1个疑似致病突变:c.10678G>A(p.G3560R)杂合突变,先证者妻子未检测到该突变。先证者母亲及姐姐均携带有PKD1基因c.10678G>A突变,而经影像学检查明确诊断为PDK且伴有典型ADPKD临床表现的先证者弟弟未检测到该突变。有相关文献[19-20]报道认为该突变可能为不致病突变或多态位点。结合本研究家系中各成员临床表现和已有相关文献报道,认为该突变位点致病可能性小,不是导致该家系胎儿多囊肾的原因。先证者弟弟为临床明确诊断的成人型PKD患者,该患者拒绝行全面的遗传性肾病高通量测序基因筛查,目前该患者病因尚不明确。
通过现有影像学及高通量测序筛查技术,大部分PKD患者都可以明确病因,但仍有少部分患者不能确定遗传学病因[6]。本研究家系中同时有两次多囊肾胎儿孕育史、成人型PKD患者史及多位家族成员肾脏多发囊肿表现,鉴于ADPKD和ARPKD的临床异质性,考虑这两类遗传性肾病在该家系同时存在的可能性,建议该家系相关成员定期行肾脏超声和肾功能检查,同时先证者妻子在孕期详细行胎儿肾脏超声检查以降低该家庭的生育风险。
