1例肝豆状核变性合并杜兴型肌营养不良症及其家系报道
2019-05-17闫建国陈大为徐志强王丽旻王福川
闫建国,陈大为,董 漪,徐志强,王丽旻,甘 雨,王福川, 张 敏
肝豆状核变性是一种遗传性铜代谢障碍疾病,由人13号染色体ATP7B基因突变造成铜沉积引起肝脏和神经系统相应症状,发生率约1∶10 000~1∶30 000。杜兴型肌营养不良症(Duchenne muscular dystrophy, DMD)是一种X染色体隐性遗传疾病,主要发生于男孩,新生男婴发生率约1∶3500。该病主要表现为骨骼肌不断退化出现肌肉无力或萎缩,逐渐丧失行走能力,通常到20多岁因心肌、肺肌无力而死亡。患者同时并发这两种疾病的概率极低,其出现可能与近亲通婚等因素有关。本文报道1例确诊为DMD合并肝豆状核变性的罕见病例,并对其进行了家系调查。
1 病例报告
1.1 病史 先证者,男,7岁,因“发现转氨酶高3年余”就诊于中国人民解放军总医院第五医学中心。4岁时体检发现肝功能异常,间断复查,仍异常(ALT 70~200 U/L),同时伴运动能力发育差,行走时步态异常,走路易摔倒,且状况逐渐加重。患儿系第2胎,足月顺产。
1.2 入院查体 患儿全身皮肤、巩膜无黄染,未见瘀斑、出血点。肝掌阳性,蜘蛛痣阴性。头颅五官无畸形,甲状腺无肿大,心肺阴性。全腹无压痛、反跳痛,肝脾肋下未触及。走路步态不稳,翼状肩胛,双侧腓肠肌肥大。肌力检查:颈曲2级,上肢内收3级,下肢内收、伸膝2级,屈髋、背伸3级,跟腱挛缩,腱反射减低。Gowers征阳性。
1.3 实验室检查及其他辅助检查 实验室检查:ALT 386 U/L、AST 231 U/L、CK 9288 U/L、CKMB 207.4 ng/ml、铜蓝蛋白 0.02 g/L、24 h尿铜105.8 μg(参考值 15.0~ 30.0 μg)。腹部超声:肝回声增粗、脾大。眼科检查:双眼角膜K-F环阴性。心脏超声:三尖瓣少量反流。肝脏穿刺病理检查:考虑遗传代谢障碍性肝病,肝豆状核变性可能性大,Scheuer肝组织学评分G2S2-3;铜染色(+)。MRI:头颅未见异常;双侧臀大肌及大腿肌肉脂肪浸润伴多发水肿改变。左肱二头肌病理诊断:骨骼肌呈肌营养不良样病理改变。
1.4 基因检测 全外显子基因检测:患儿存在2处肝豆状核变性相关基因ATP7B纯合突变(ATP7B c.2333G>T chr13:52532469 p.R778L纯合突变——已知致病突变;ATP7B c.2310C>G chr13:52532492 p.L770L纯合突变——疑似致病突变),家系验证结果显示此2处纯合突变分别来自其父母(图1)。同时发现该患儿DMD基因51号外显子有缺失突变。线粒体全基因检测未发现异常。全外显子基因检测未见明确大片段缺失及重复突变。
1.5 病例诊断 根据患儿临床表现、查体、生化指标、辅助检查及基因检测,确诊为DMD合并肝豆状核变性。
2 家系调查
通过详细问诊,进行家系上溯4代调查,发现有近亲结婚及多例肝豆状核变性和/或DMD患者。此外,行父亲、母亲、姐姐、表兄、小姨、舅舅基因验证,ATP7B c.2333、ATP7B c.2310、DMD基因51号外显子的3个结果如图1~3所示。
先证者父母为近亲结婚[先证者的奶奶(Ⅱ-2)与其姥爷(Ⅱ-8)是亲兄妹]。基因验证结果显示先证者母亲(Ⅲ-3)存在ATP7B基因双杂合突变及DMD基因51号外显子的杂合缺失变异;先证者父亲(Ⅲ-2)存在ATP7B基因双杂合突变,无DMD基因51号外显子的缺失或重复变异,临床表型正常;先证者的姐姐(Ⅳ-1)与先证者存在相同位点的ATP7B基因纯合突变,临床诊断为肝豆状核变性,同时存在DMD基因51号外显子的杂合缺失变异,为DMD女性携带者。
先证者大姨(Ⅲ-4)是其母亲(Ⅲ-3)的同卵双生姐姐(外观推测,未行基因验证)。其儿子[先证者表哥(Ⅳ-3)]亦转氨酶高,且伴肌酸激酶显著升高,有DMD基因51号外显子缺失突变,未发现ATP7B基因突变,确诊为DMD。
先证者舅舅(Ⅲ-7)未发现ATP7B基因突变及DMD基因51号外显子的缺失或重复变异;其小姨(Ⅲ-6)存在ATP7B基因双杂合突变,未见DMD基因51号外显子的缺失或重复变异,临床表型正常。
先证者母系Ⅱ代亲属中有3人约50岁死于不明原因肝硬化;推测II-8、II-9均携带ATP7B突变;II-9携带DMD基因,且III-3、III-4的DMD基因异常均源自II-9;推测II-9家族有DMD和ATP7B 2个致病基因,I-4女性为两病携带者或DMD携带+肝豆状核变性患者。根据问诊及基因验证画出家系图谱(图4)。
3 讨 论

图1 ATP7B c.2333G>T二代测序图a. 先证者;b. 先证者母亲;c. 先证者父亲;d. 先证者姐姐;e. 先证者小姨;f. 先证者舅舅Figure 1 ATP7B c.2333G>T next generation sequencing

图2 ATP7B c.2310C>G二代测序图a. 先证者;b. 先证者母亲;c. 先证者父亲;d. 先证者姐姐;e. 先证者小姨;f. 先证者舅舅Figure 2 ATP7B c.2310C>G next generation sequencing
肝豆状核变性,又称Wilson病,是一种常染色体隐性遗传疾病,是由编码铜转运所需P型ATP酶的ATP7B基因突变所致。病理学改变首先表现为肝脏中铜的毒性蓄积,铜过载后便在其他部位(例如神经系统、角膜、肾脏和心脏)中沉积,逐步进展为肝硬化、肝功能衰竭或不可逆的脑损伤。肝豆状核变性的诊断主要通过评分系统确诊(铜蓝蛋白、24 h尿铜定量、肝铜、罗丹宁铜染色、K-F环、神经系统症状、coomb’s阴性的溶血、基因检测)[1-2]。目前,国内研究认为Arg778Leu是中国人肝豆状核变性的突变热点[3]。本例先证者经基因检测证实,存在2处纯合突变,为ATP7B c.2333G>T chr13:52532469 p.R778L(已知致病突变)和c.2310C>G chr13:52532492 p.L770L(疑似致病突变),其父母、小姨、姐姐均存在相同突变位点,但因为双杂合不发病。结合家族史及家系图谱,可预知父系和母系Ⅰ代亲属及Ⅱ代直系亲属中均存在ATP7B基因的相同突变位点,且先证者母系Ⅱ代亲属中Ⅱ-11、Ⅱ-12、Ⅱ-13均因肝硬化约于50岁死亡,肝豆状核变性可能性大。先证者家系中死于肝病者均已到成年,推测先证者及其姐姐肝豆状核变性病情进展可能缓慢。
假肥大型肌营养不良症包括DMD和贝克型肌营养不良症(Becker muscular dystrophy, BMD),二者均是由于抗肌萎缩蛋白(dys)基因突变所致的X-连锁隐性遗传病。男婴DMD的发病率约为30/10万。DMD/BMD患者dys缺乏会引起骨骼肌细胞膜缺陷,细胞内的肌酸激酶等外漏,肌细胞坏死,脂肪组织和纤维结缔组织增生[4]。DMD早期的主要表现为下肢近端和骨盆带肌萎缩和无力、小腿腓肠肌假性肥大、鸭步和Gowers征,晚期可出现全身骨骼肌萎缩,通常在20多岁死于呼吸衰竭或心力衰竭。DMD患者病情进展快,预后差。通过典型临床表现(双下肢无力进行性发展、腓肠肌肥大)、肌酶显著升高、肌电图、肌活检、超声心动图、肌肉MRI、DMD基因检测进行确诊[5]。对于典型的DMD患儿,若基因检测已确诊,则不须再行肌活检与肌电图。本例先证者经基因检测证实DMD基因51号外显子存在缺失突变;其母亲、姐姐均存在相同位点的杂合缺失变异,但运动能力正常,检测肌酶正常,未诊断为DMD,但均为DMD携带者;其母亲及其母亲的孪生姐姐为同卵双生,其表哥(Ⅳ-3)存在DMD基因51号外显子缺失突变,确诊DMD。结合家族史及家系图谱,可预知先证者母亲及其母亲的孪生姐姐(Ⅲ-4)再次生育男婴患DMD概率高达50%。而经基因检测,Ⅲ-6、Ⅲ-7男、女各1例均未见DMD基因51号外显子的缺失变异,Ⅰ代亲属及Ⅱ代直系亲属男女均无DMD家族史,考虑DMD基因变异来自Ⅰ-4及Ⅱ-9女性携带者。可进一步完善Ⅱ代基因检测以证实。

图3 DMD基因51号外显子测序图a. 先证者;b. 先证者母亲;c. 先证者姐姐;d. 先证者小姨;e. 先证者舅舅;f. 先证者表哥Figure 3 Sequencing of exon 51 of DMD gene
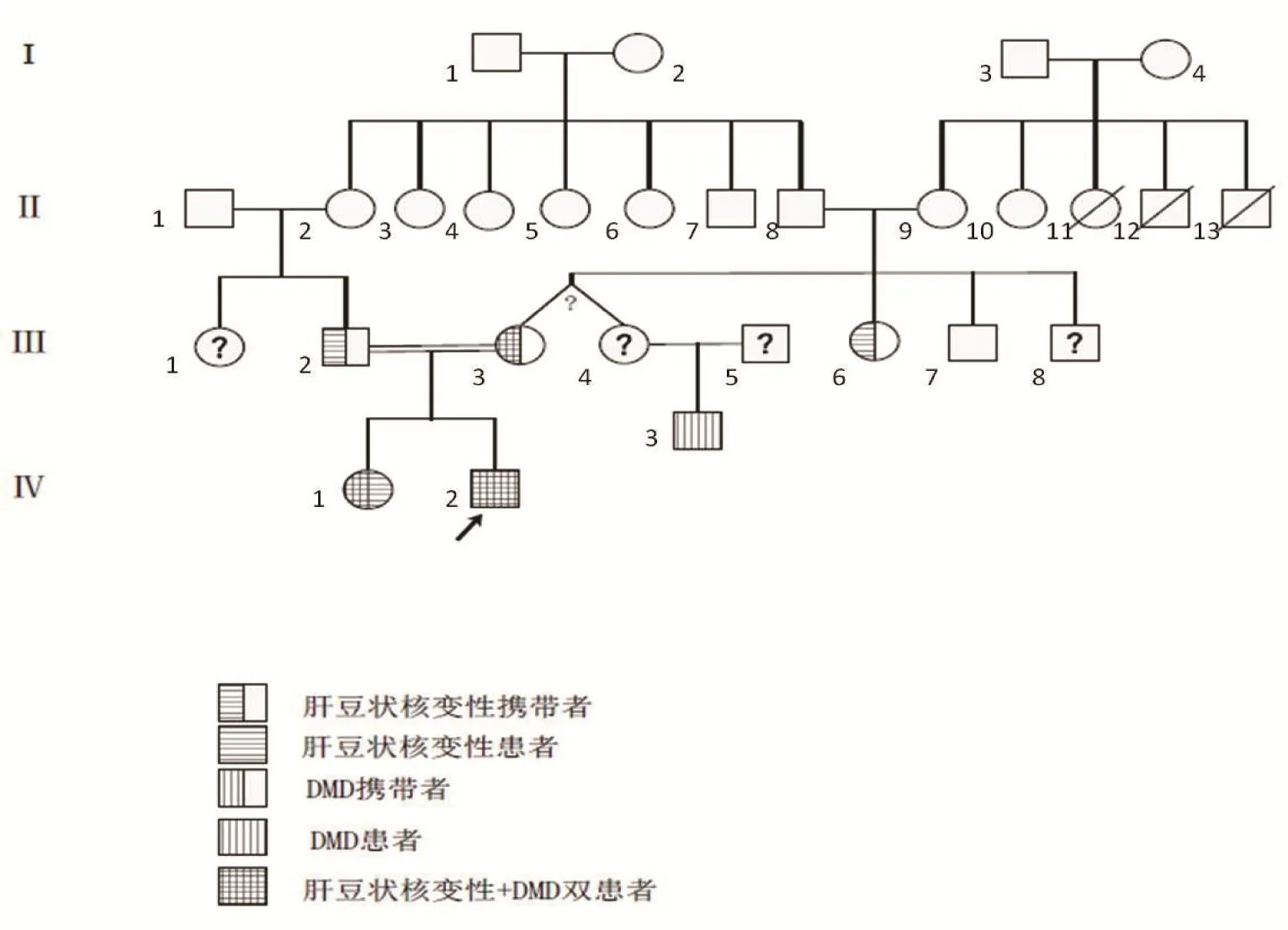
图4 家系图谱Figure 4 Family genetic atles
另外,通过家系分析可知,Ⅰ代父系家庭中含有ATP7B致病突变,Ⅰ代母系超家族中含有ATP7B致病突变和女性DMD基因51号外显子缺失突变携带,Ⅱ代母系家族已出现多名肝豆状核变性患者。Ⅲ代母系家族中2名女性携带DMD基因51号外显子缺失突变,与Ⅲ代携带ATP7B致病突变的男性近亲通婚,导致Ⅳ代男性出现肝豆状核变性合并DMD。因此,Ⅳ代女性为DMD基因携带者,生育男婴患DMD概率高达50%。为防止Ⅳ代女性子女的发病,强烈建议进行遗传咨询及产前检查。
肝豆状核变性与DMD均为罕见遗传病,国内几乎未见同时患有两种疾病的报道。肝细胞损伤及肌细胞损伤均可导致转氨酶的升高,转氨酶升高的鉴别诊断对于治疗意义重大。本报道通过对病史、临床表现、体格检查、辅助检查及影像学、病理学、基因检测的总结分析,最终才获得确诊。由于DMD尚无有效治疗方法,故应以预防为主。而高效、准确的基因检测手段有助于进行遗传咨询及产前诊断,避免患病儿出生,为预防本病提供关键的技术支持[6]。随着基因编辑技术的快速进展,在基因诊断的基础上进行有效的基因治疗成为可能,为先天遗传性疾病的治疗带来希望[7-9]。
