miR-145过表达对缺氧诱导心肌细胞焦亡的抑制作用及其机制
2022-11-15浦湧崔胜宇吴浩亮陶波孟祥平夏豪徐林
浦湧,崔胜宇,吴浩亮,陶波,孟祥平,夏豪,徐林
1武汉经济技术开发区(汉南区)人民医院武汉大学人民医院汉南医院心内科,武汉 430090;
2武汉大学人民医院武汉大学心血管病研究所心血管病湖北省重点实验室心内科;
3武汉市第四医院老年医学科
微小RNA(miRNA)是一种小的非编码RNA,可与相关靶基因的3'-非翻译区结合,对靶基因产生负调控作用[1]。miRNA广泛参与细胞增殖[2]、凋亡[3]、分化[4]等生理或病理过程,在细胞内具有重要的调控作用。作为miRNA的一种,miR-145已被证明对心血管疾病具有至关重要的影响。焦亡是细胞程序性死亡的一种类型,其在各种心血管疾病的发病中起着关键作用,可发生于心肌梗死[5]、心肌缺血再灌注损伤[6]、动脉粥样硬化[7]、心力衰竭[8]等疾病中。焦亡同凋亡类似,是一个高度调控的细胞死亡过程,通过多种方式干预抑制这一过程在许多条件下具有心脏保护作用。炎症小体的激活、Caspase-1的活性转化、活性氧(ROS)表达增加以及OD样受体热蛋白结构域相关蛋白3(NLRP3)、(IL-1β)、(IL-18)等炎症因子的高表达是细胞焦亡发生的关键特征[9]。研究发现,miR-145能够靶向负调控钙调蛋白依赖性蛋白激酶Ⅱ(CaMKⅡ),进而调控其下游分子核转录因子κB(NF-κB)[10],且NF-κB信号的激活可进一步调控NLRP3的激活[11]。由于miR-145对心血管疾病具有的潜在保护作用,并且焦亡也发生于心肌细胞的损伤过程中,因此有必要研究miR-145是否可能通过对心肌细胞焦亡的调控发挥对心肌细胞的保护作用,同时是否通过CaMKⅡ/NF-κB信号通路影响心肌细胞焦亡。2022年1月—6月,本研究通过缺氧诱导心肌细胞损伤,观察miR-145过表达对心肌细胞焦亡的影响并分析其机制。现报告如下。
1 材料与方法
1.1 主要材料大鼠心肌细胞系H9C2购自中国科学院上海细胞库,胎牛血清购自美国BIOEXPLORER Life Science公司,高糖培养基购自美国Cytiva公司,0.25%胰蛋白酶购自美国Gibco公司,过表达miR-145腺病毒及其阴性对照病毒购自上海吉凯公司。超氧阴离子检测试剂(活性氧检测试剂)购自碧云天生物公司,逆转录-PCR试剂盒购自赛维尔生物科技公司,AnnexinV-PE/7-AAD凋亡染色试剂盒购自美国BD公司。磷酸化CaMKⅡ(p-CaMKⅡ)、磷酸化NF-κB(p-NF-κB)、GAPDH抗体均购自美国CST公司,NLRP3、Caspase-1、IL-1β、IL-18山羊抗兔及山羊抗小鼠抗体购自上海艾比玛特生物医药公司。
1.2 实验方法
1.2.1 细胞培养及分组H9C2细胞置于含10%胎牛血清的高糖培养基中,于37℃、5% CO2的细胞培养箱中培养。待细胞融合度达70%~80%时,分为正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组。
1.2.2 细胞miR-145腺病毒感染及缺氧诱导缺氧+miR-145组、缺氧+阴性对照组分别使用腺病毒包装的miR-145及腺病毒包装的阴性对照序列与细胞共孵育6 h后换液,病毒感染复数(MOI)=50;正常组、缺氧组不做miR-145腺病毒感染处理。24 h后将缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞转移至缺氧培养箱(1%氧气浓度)中培养,正常组不做缺氧处理,24 h后收集细胞进行后续实验。
1.2.3 细胞miR-145 mRNA表达检测采用实时荧光定量PCR法。收集各组细胞,按照试剂盒说明常规提取总RNA,而后逆转录为cDNA,以此为模板,按照实时荧光定量PCR的说明书进行荧光定量PCR。循环条件:95℃10 min,95℃15 s、60℃30 s,共完成40个循环。引物序列:miR-145-5p上游为5′-ACACTCCAGCTGGGGTCCAGTTTTCCCAGGA-3′,下 游 为5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGGGATTC-3′;U6上游为5′-CTCGCTTCGGCAGCACA-3′,下 游 为5′-AACGCTTCACGAATTTGCGT-3′。以U6为内参,按照用2-ΔΔCt计算各组miR-145的相对表达量。
1.2.4 细胞ROS水平检测采用超氧化物阴离子探针(碧云天,S0063)。使用终浓度为2.5 μmol/L的超氧化物阴离子探针和细胞一起在37℃孵育30 min进行荧光探针装载,PBS洗涤后,使用荧光显微镜观察拍照。
1.2.5 细胞焦亡情况观察采用TUNEL染色及流式细胞术。TUNEL染色:将各组细胞与TUNEL反应液在37℃黑暗条件下孵育90 min,用荧光显微镜记录并拍照,计数TUNEL阳性细胞以反映心肌细胞焦亡情况。TUNEL阳性率=TUNEL阳性细胞数/总细胞数×100%。流式细胞术:取各组细胞,收集原培养液,PBS洗后加入胰酶消化1~3 min后终止消化,用移液枪吹打成单个细胞与上清液混匀,1 000 r/min离心5 min,去上清,PBS漂洗,1 000 r/min离 心5 min,去上清。加入100 μL 1X Annexin Binding Buffer重悬细胞,加入5 μL Annexin V-PE,加入5 μL 7-AAD,室温避光15 min,上机前补加400 μL 1X Binding Buffer,上流式细胞仪检测各组细胞死亡率。
1.2.6 细胞中焦亡相关因子表达检测采用免疫组织化学及实时荧光定量PCR法。采用免疫组化法检测NLRP3蛋白表达:细胞样本在4%多聚甲醛固定后,使用内源性过氧化物酶阻断剂孵育20 min,后漂洗3次,使用3%BSA封闭样本3 min后,滴加配置好的NLRP3一抗液体孵育过夜。第2天将玻片孵育二抗后,使用DAB显色,复染细胞核封片后在光镜下拍照,观察NLRP3蛋白表达情况。采用实时荧光定量PCR法检测NLRP3、IL-1β、IL-18 mRNA表达:方法同“1.2.3”,循环条件:95℃30 s,95℃15 s、60℃10 s,共完成40个循环。引物序列:NLPR3上游 为5′-AGCTGCTCTTTGAGCCTGAG-3′,下 游 为5′-TCTGCTAGGCTCTTTGGTGC-3′;IL-1β上 游 为5′-TCGTGCTGTCTGACCCATGT-3′,下 游 为5′-ACAAAGCTCATGGAGAATACCACTT-3′;IL-18上游为5′-ACGGAGCATAAATGACCAAGTTC-3′,下游为5′-TCTGGGATTCGTTGGCTGTT-3′;GAPDH上游为5′-CCAAGGTCATCCATGACAACTT-3′,下游为5′-AGGGGCCATCCACAGTCTT-3′。以GAPDH为 内参,按 照 用2-ΔΔCt计 算 各 组NLRP3、IL-1β、IL-18 mRNA的相对表达量。
1.2.7 细胞中CaMKⅡ/NF-κB信号通路相关蛋白表达检测采用Western blotting法。取各组细胞在裂解缓冲液中裂解得到总蛋白,通过凝胶电泳分离转移到PVDF膜上。在5%的脱脂牛奶中封闭60 min后,在4℃与p-CaMKⅡ、p-NF-κB、NLRP3、Caspase-1、IL-1β、IL-18及GAPDH抗体孵育过夜。第二天使用缓冲液洗膜后,在室温下与辣根过氧化物酶标记的山羊抗兔二抗或抗小鼠二抗孵育1 h。利用增强型化学光检测试剂对蛋白条带进行可视化,利用Image J软件分析条带灰度值。以GAPDH作为内源性对照,以目的条带与对照条带灰度比值作为目的蛋白相对表达量。
1.3 统计学方法采用SPSS23.0统计软件。计量资料采用Shapiro-Wilk方法行正态性检验,符合正态分布且方差齐性的计量资料以±s表示,多组间比较采用单因素方差分析,两组比较采用t检验。P<0.05为差异有统计学意义。
2 结果
2.1 各组细胞miR-145 mRNA表达比较正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞miR-145 mRNA相对表达量分别为(1.00±0.10)、(0.62±0.05)、(6.43±0.40)、(0.59±0.14),细胞miR-145 mRNA相对表达量缺氧+miR-145组>正常组>缺氧组、缺氧+阴性对照组(P均<0.05)。
2.2 各组细胞ROS水平比较正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞ROS水平分 别 为(1.04±0.09)、(3.00±0.20)、(1.61±0.17)、(3.25±0.23),细胞ROS水平缺氧组、缺氧+阴性对照组>缺氧+miR-145组>正常组(P均<0.05)。
2.3 各组细胞中焦亡情况比较TUNEL染色显示,正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞TUNEL阳性率分别为(14.33±4.04)、(60.00±3.00)、(45.00±3.00)、(65.00±2.29);流式细胞术检测显示,正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞死亡率分别为(4.30±0.26)、(14.33±0.44)、(9.20±0.51)、(14.46±0.35);细胞TUNEL阳性率、死亡率缺氧组、缺氧+阴性对照组>缺氧+miR-145组>正常组(P均<0.05)。
2.4 各组细胞中焦亡相关因子表达比较免疫组化染色显示,正常组、缺氧组、缺氧+miR-145组、缺氧+阴性对照组细胞NLRP3蛋白表达分别为(1.00±0.20)、(5.43±0.21)、(2.70±0.26)、(5.73±0.21),细胞NLRP3蛋白表达缺氧组、缺氧+阴性对照组>缺氧+miR-145组>正常组(P均<0.05)。实时荧光定量PCR显示,NLRP3、IL-1β、IL-18 mRNA表达缺氧组、缺氧+阴性对照组>缺氧+miR-145组>正常组(P均<0.05)。见表1。
表1 各组NLRP3、IL-1β、IL-18 mRNA表达比较(±s)
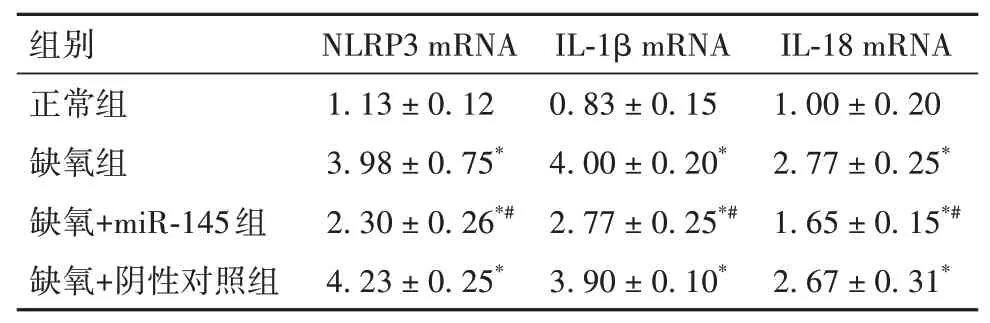
表1 各组NLRP3、IL-1β、IL-18 mRNA表达比较(±s)
注:与正常组比较,*P<0.05,与缺氧组比较,#P<0.05。
?
2.5 各组中CaMKⅡ/NF-κB信号通路相关蛋白表达比较细胞p-CaMKⅡ、p-NF-κB、NLRP3、Caspase-1、IL-1β、IL-18蛋白表达缺氧组、缺氧+阴性对照组>缺氧+miR-145组>正常组(P均<0.05)。见表2。
表2 各组CaMKⅡ/NF-κB信号通路相关蛋白表达比较(±s)
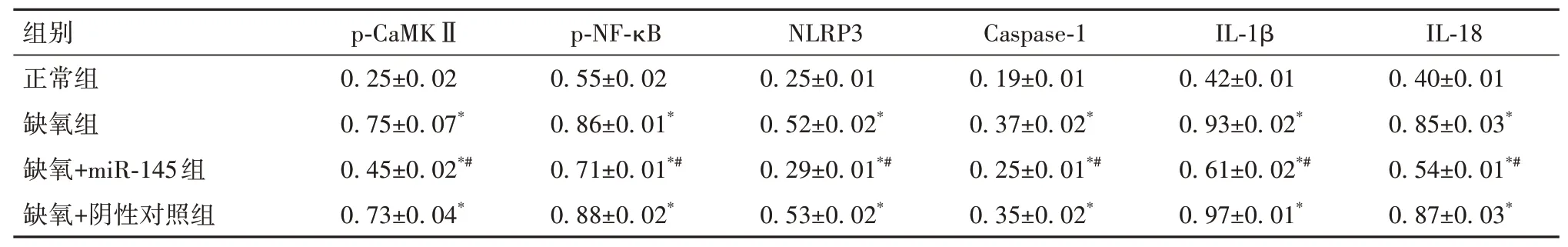
表2 各组CaMKⅡ/NF-κB信号通路相关蛋白表达比较(±s)
注:与正常组比较,*P<0.05,与缺氧组比较,#P<0.05。
?
3 讨论
心肌梗死是一种严重的冠状动脉相关疾病,主要由冠状动脉粥样硬化血栓形成或多种原因造成心肌氧供失衡、心肌细胞损伤导致。动脉粥样硬化斑块被破坏后,引起血小板聚集,导致冠状动脉阻塞,心肌缺血缺氧坏死。研究表明,梗死的心肌及缺血缺氧的心肌均伴随细胞焦亡,发生梗死的心肌细胞中NLRP3、IL-1β、IL-18等炎症细胞因子水平普遍升高,而施加干预对NLRP3炎症小体抑制后,可以减少心肌焦亡,从而有效改善缺血缺氧诱导的心肌损伤[12]。miR-145定位于染色体5q32-33,长度约为4.08 kb,已被证明在多种心血管疾病中有着积极影响,并且可在心肌梗死相关损伤中发挥重要的调控作用。我们的研究发现,在遭受缺氧的H9C2心肌细胞中,miR-145表达显著下调,这与LIU等[13]研究结果一致。而通过miR-145腺病毒感染H9C2细胞,发现细胞中miR-145表达显著上调,提示成功建立过表达miR-145的腺病毒感染细胞。
研究显示,在缺血缺氧的心肌中,过度的炎症、氧化应激水平会加重心肌细胞损伤[10]。我们在研究中检测了各组细胞内的ROS水平,发现缺氧组及缺氧+阴性对照组细胞内ROS水平较正常组均明显升高,而缺氧+miR-145组细胞内ROS水平较缺氧组及缺氧+阴性对照组明显下降,提示miR-145过表达的缺氧心肌细胞内ROS水平降低,氧化应激水平得到明显改善。
细胞焦亡是与炎症密切相关的程序性死亡,我们在研究中进一步检测了细胞焦亡相关情况,发现经过缺氧诱导的细胞TUNEL阳性率、流式细胞术检测的细胞死亡率均高于正常组,而在过表达miR-145的细胞中,细胞TUNEL阳性率、死亡率较缺氧组及缺氧+阴性对照组均有明显改善。此外,我们还对焦亡发生的特征性指标NLRP3蛋白进行了免疫组化染色,并且对焦亡相关效应分子NLRP3、IL-1β、IL-18 mRNA进行了实时荧光定量PCR检测,发现在缺氧+miR-145组中,NLRP3、IL-1β、IL-18表达均低于缺氧组及缺氧+阴性对照组,提示在心肌细胞中过表达miR-145可减轻缺氧引起的细胞焦亡。
ROS可激活CaMKⅡ信号进而加重心肌细胞损伤,并且miR-145被发现可通过对CaMKⅡ的靶向负调控发挥调控其下游信号的作用[13-14]。CaMKⅡ是丝氨酸/苏氨酸蛋白激酶家族成员之一,广泛参与细胞钙信号转导、细胞存活、细胞增殖以及细胞炎症或氧化应激反应[15-16]。目前,关于CaMKⅡ对细胞焦亡过程影响的报道较少,但CaMKⅡ在细胞焦亡过程中的可能作用仍可从以往的相关研究中推测。SUETOMI等[17]发现,心肌细胞中缺失CaMKⅡ可降低压力过载诱导的心功能障碍和心脏炎症水平,这种保护作用可能与缺失CaMKⅡ抑制了NF-κB及NLRP3的激活有关。此外,一项关于CaMKⅡ介导胶质母细胞瘤细胞中焦亡的研究显示,Caspase家族及GSDME主要参与了CaMKⅡ介导的焦亡[18]。NFκB信号可作为CaMKⅡ的下游分子发挥作用[19],并且有研究报道,激活的NF-κB信号可进一步调控NLRP3的激活[11]。本研究采用Western blotting法检测了CaMKⅡ/NF-κB信号通路相关蛋白表达,发现在过表达miR-145的心肌细胞中,由缺氧引起的过度激活的CaMKⅡ信号被抑制,具体表现为p-CaMKⅡ较缺氧+阴性对照组显著下调。此外,我们通过观察NF-κB及其下游的NLRP3介导的焦亡信号通路发现,在缺氧及缺氧+阴性对照组中,CaMKⅡ/NF-κB信号通路及其下游的焦亡指标NLRP3、Caspase-1、IL-1β、IL-18均被激活;而在缺氧+miR-145组中,这些指标均有了显著改善。以上结果提示,miR-145可以缓解ROS的产生,并且可抑制CaMKⅡ的活性进而降低p-NF-κB水平,影响NF-κB核转位,这一级联效应使焦亡相关蛋白NLRP3、Caspase-1、IL-1β和IL-18的表达受到抑制,最终减轻了心肌细胞焦亡。
综上所述,本研究通过miR-145腺病毒感染心肌细胞系H9C2并对其进行缺氧诱导,发现miR-145可通过对CaMKⅡ/NF-κB信号通路的抑制来发挥对心肌细胞的抗焦亡作用,进而保护缺氧引起的心肌细胞损伤。尽管miR-145在心血管疾病中的保护作用已被广泛证实,但其对焦亡的调控作用尚未见报道,本研究或可为心肌损伤今后可能的靶向治疗以及miR-145的生物学功能提供新的见解。
