MBTPS2基因新发突变致毛囊性鱼鳞病-脱发-畏光综合征一例
2022-08-20吕小岩
郑 铭 吕小岩
1 四川大学华西医院皮肤科,成都,610041;2 四川大学华西医院疾病分子网络前沿科学中心免疫炎症研究院皮肤病学研究室,成都,610041
毛囊性鱼鳞病-脱发-畏光综合征(ichthyosis follicularis, atrichia and photophobia syndrome, IFAP)由Macleod[1]于1909年首次提出,表现为不同程度的毛囊性鱼鳞病、脱发、畏光,是一种罕见的遗传性疾病。现将我科收治的1例IFAP报道如下。
临床资料患儿,男,4岁。患儿出生时头发、眉毛缺如,全身泛发粟粒大毛囊性角化过度性丘疹,以头皮、项背部为重。患儿4个月大时,家属发现其畏光,当地医院诊断为“结膜炎”,予以治疗(具体不详)后未见明显好转。既往史、出生史、喂养史、家族史等无特殊。
体格检查:身高90.5 cm,体重13.5 kg,智力发育正常,畏光,余各系统未见异常。皮肤科检查:全身皮肤干燥,可见粟粒大毛囊性角化过度性丘疹,呈钉突状,荆棘样触感,眉毛缺失,头皮仅可见稀疏毛发,无瘢痕(图1)。实验室检查:血常规、肝肾功能、甲状腺功能、梅毒、艾滋病等未见异常。
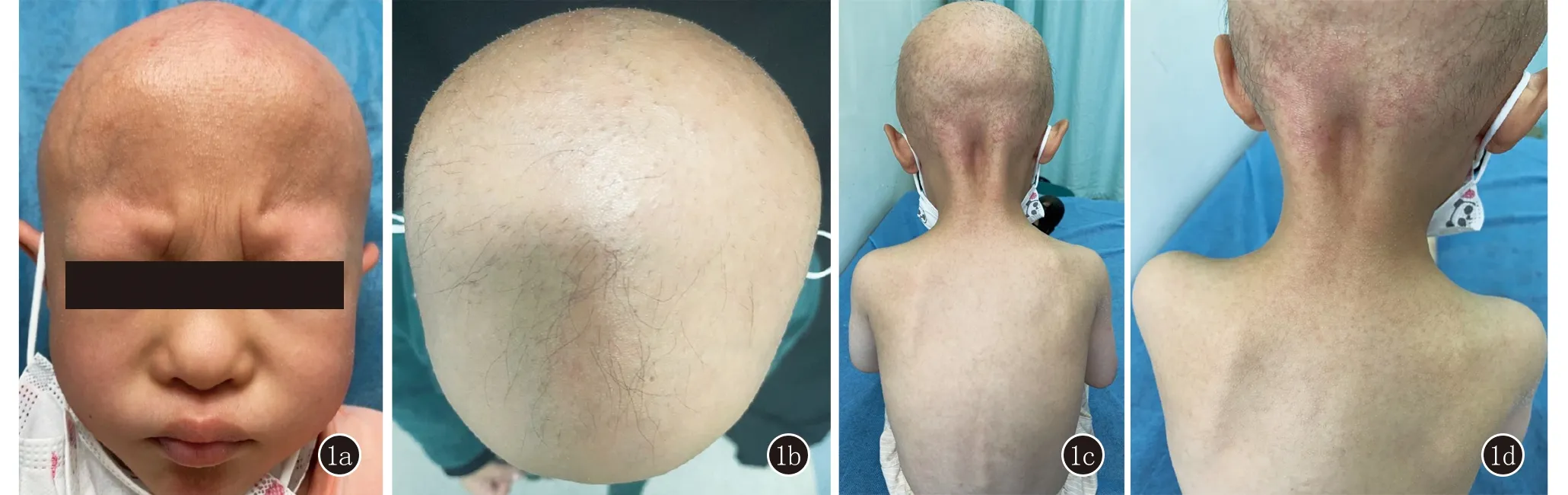
图1 1a、1b:头皮非瘢痕性脱发,眉毛缺如,睫毛存在;1c、1d:全身泛发毛囊性角化过度性丘疹,头皮、肩背部较重
基因检测:患儿MBTPS2基因有1个半合子突变,3号核苷酸由鸟嘌呤G变为胞嘧啶C,导致第1号氨基酸由甲硫氨酸变为异亮氨酸,即c.3G>C(p.M1I)。父亲该位点无变异,母亲该位点杂合变异(图2)。
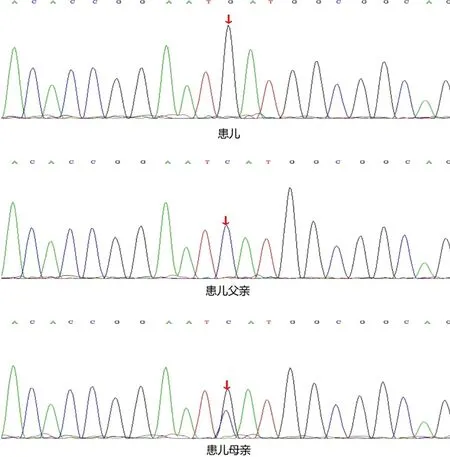
图2 患儿MBTSP2基因存在1个半合子突变c.3G>C(p.M1I),父亲该位点无变异,母亲为杂合子突变
诊断:毛囊性鱼鳞病-脱发-畏光综合征。
讨论IFAP是一种罕见的遗传性疾病,其最常见的皮肤表现为非炎症性毛囊性角化过度性丘疹,主要累及头皮和四肢伸侧。IFAP另一典型的皮肤表现是先天性非瘢痕性非炎症性脱发,可累及睫毛、眉毛,亦有患者仅表现头皮毛发稀疏[2],也有后天性脱发的报道[3]。此外,畏光也是IFAP的诊断依据之一。IFAP还有一系列伴随症状,如甲营养不良、癫痫发作、生长发育迟缓、智力发育落后、特应性皮炎、隐睾等。本例患儿以皮肤、毛发和眼部受累为主,生长发育落后,尚未有其他系统受累表现。
IFAP多数为男性患者,亦有女性IFAP患者被报道[4]。Oeffner等[5]首次发现IFAP的发病与X染色体p22.11-p22.13片段上的MBTSP2基因突变有关,提示本病可能主要呈X连锁隐性遗传。MBTSP2基因可编码膜结合转录因子蛋白酶位点2(membrane-bound transcription factor protease site-2,MBTPS2),MBTPS2可以激活固醇调节元件结合蛋白,对于维持正常皮肤屏障的功能具有重要作用,MBTPS2的受损程度与IFAP疾病严重程度相关[5,6]。Jiang等[7]总结了与IFAP相关的MBTSP2基因突变,包括c.261G>A、c.1286G>A、c.667G>T、c.1424T>C、c.225‐6T>A、c.1523A>G、c.1433C>A、c.671‐9T>G、c.1001G>A、c.71T>C、c.774C>G、c.758G>C、c.686T>C、c.1427T>C、c.1430A>T、c.1499G>A、c.1538T>C、c.1523A>C、c.1360G>C。本例患者MBTSP2基因c.3G>C(p.M1I),为全球首报。
MBTSP2基因突变亦与脱发性小棘毛囊角化病(keratosis follicularis spinulosa decalvans,KFSD)、X连锁Olmsted综合征有关,因此需要与本病进行鉴别。KFSD主要表现为泛发性毛囊性角化过度性丘疹、头皮瘢痕性脱发、面部红斑等[8],而本例患者表现为非瘢痕性脱发。X连锁Olmsted主要特征为对称性残毁性掌跖角皮症、口周角化过度性斑块[9],本例患者均无上述表现。Wang等[10]曾报道一例同时具有IFAP和Olmsted综合征临床表现的患者,提示MBTSP2基因突变具有临床异质性。
IFAP目前无特效疗法,外用维A酸乳膏、糖皮质激素等可以改善皮肤症状。有病例报道口服维生素A(250000单位/天),连续使用6个月可改善畏光及皮肤症状[11]。口服阿维A(1 mg/kg·d)可以使皮损变平,但对脱发及眼部症状无效[2]。Chauhan等[12]曾报道一例IFAP患者,其皮损在口服异维A酸(1 mg/kg·d)4个月后显著好转,但停药后皮损复发。上述治疗均有一定的不良反应,且异维A酸可能导致儿童骨骺盘早熟融合,使IFAP患者生长发育受限更加严重,因此在启动治疗前需要与患方充分沟通。
