毛囊角化病合并疣状肢端角化症二例临床及基因检测分析
2022-08-20周桂芝裴振环张福仁
张 帆 周桂芝 裴振环 刘 红 张福仁
山东第一医科大学附属皮肤病医院(山东省皮肤病医院),山东省皮肤病性病防治研究所,济南,250022
毛囊角化病(keratosis follicularis)又称Darier病(Darier’s disease,DD),是一种以表皮细胞角化不良为基本病理特征的角化性疾病,呈常染色体显性遗传。疣状肢端角化症(acrokeratosis verruciformis,AKV),最早在1931年由hopf报道,因此又称hopf病。典型的临床表现为手足、肘部、膝盖等部位的肤色疣状丘疹,丘疹可以散发也可聚集。手掌、足底及指甲可有充满角质的小凹陷。病理最为特异性的表现是由于角化过度形成的“教堂塔尖样”结构。DD和AKV均由ATP2A2基因突变所致,临床表现更是有很多相似之处,这使两者之间的关系受到广泛争议。我们诊治了2例疣状肢端角化症合并毛囊角化病的病例,并通过文献复习总结两者的关系。
临床资料患者1,女,11岁。因手足丘疹2年来我院就诊。患者2年前双手背部出现丘疹,伴瘙痒,后丘疹蔓延至上臂。1年来足部、腋窝、面部出现相似皮损。5个月前至当地医院就诊,以“湿疹”治疗,效果不佳,遂至我院就诊。家族史:其母及外祖母患有毛囊角化病(图1)。体格检查:系统检查未见异常。皮肤科查体:面部、腋窝、双手足背部、双上肢多发扁平肤色疣状丘疹,腋窝处丘疹覆盖油腻痂皮。指甲轻度发白,左手食指、无名指指甲可见褐色纵纹。
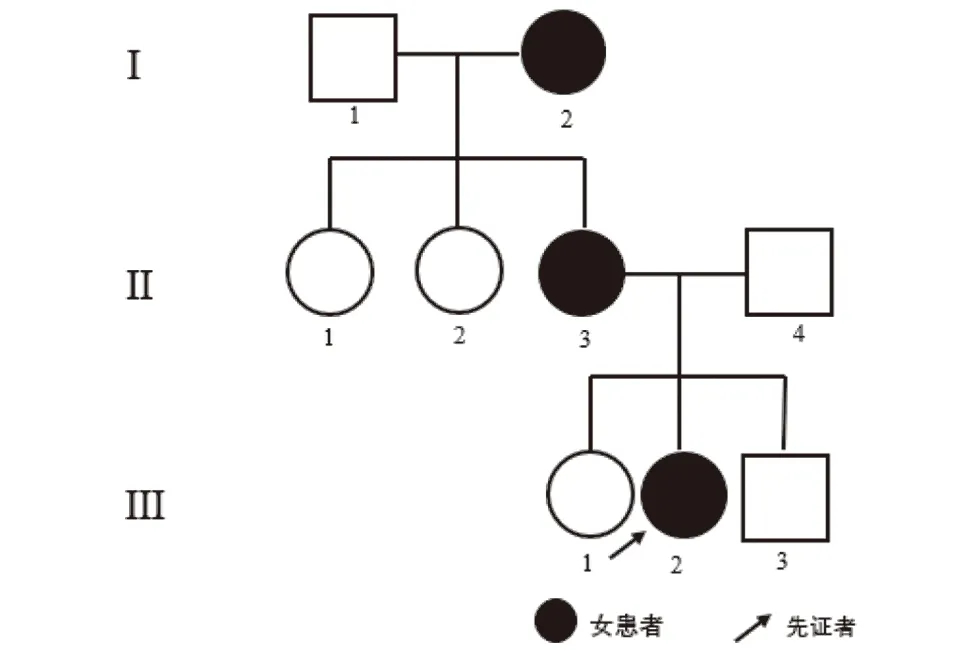
图1 患者1家系图
手掌未见点状角化(图2)。组织病理示:(左手)表皮角化过度,少许角化不全,棘层呈轻度乳头瘤状增生,真皮浅层少许单一核细胞。表皮显示出类似于教堂尖顶的独特外观(图3)。对患者本人及其父母行聚合酶链式反应(PCR)发现,患者及其母亲ATP2A2基因中15号外显子2282位均发生了G→T突变,其父ATP2A2基因未发现异常(图4)。诊断:毛囊角化病合并疣状肢端角化症。治疗:给予外用维甲酸乳膏。1个月后电话随访,患者家属诉治疗效果一般,后失访。
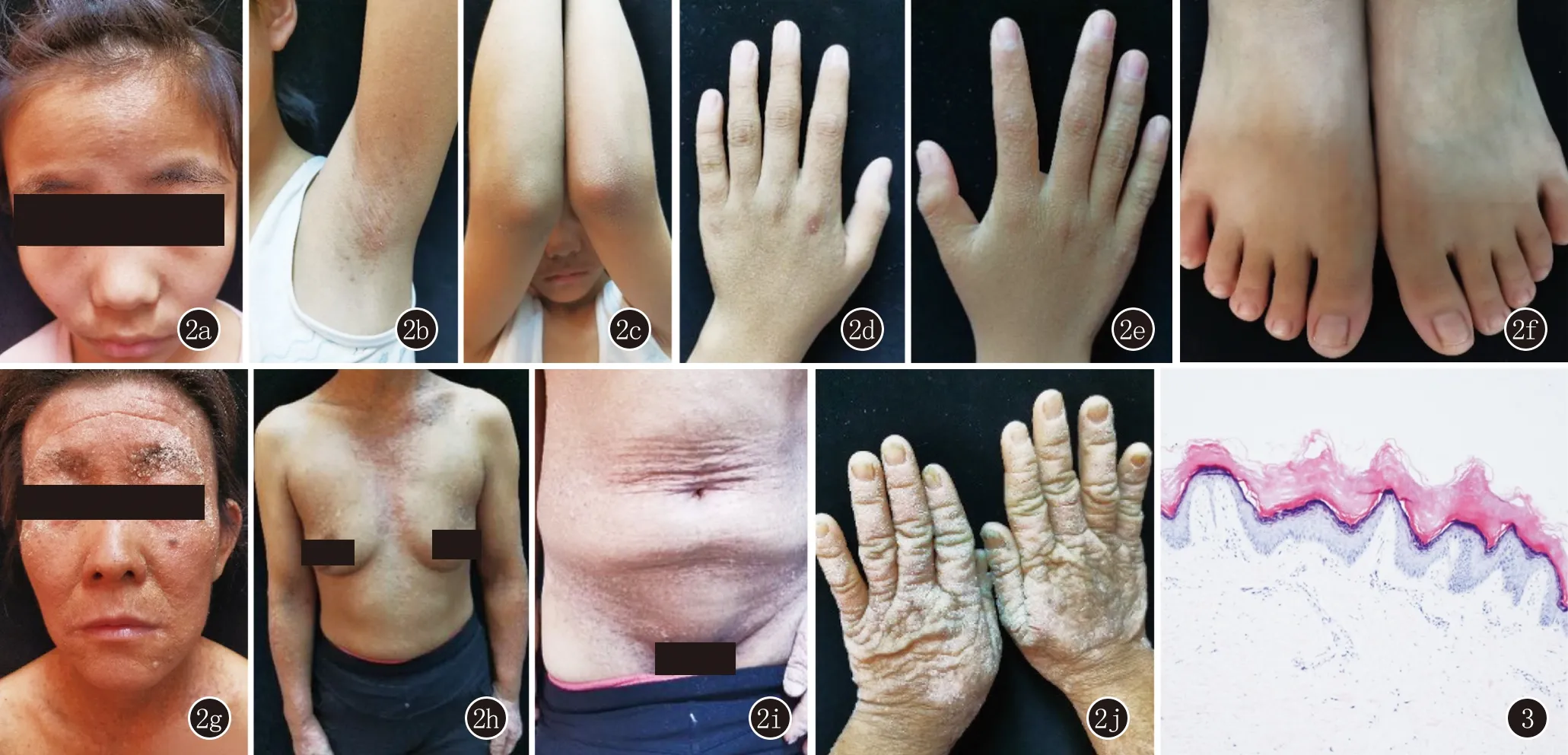
图2 2a~2g:患者肢端部位密集质硬的肤色丘疹,头面部、腋窝脂溢部位角化型丘疹,腋窝部皮损覆有油腻性痂皮;2h~2j:患者母亲面部、手部角化型丘疹融合呈疣状斑块,可见结痂,前胸、腹股沟褐色角化型丘疹 图3 表皮角化过度,少许角化不全,棘层呈轻度乳头瘤状增生,真皮浅层少许单一核细胞。表皮显示出类似于教堂尖顶的独特外观(HE,×200)
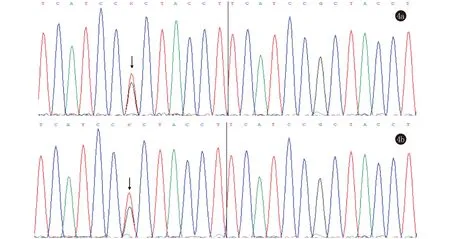
图4 4a:与正常人pcr序列对比,患者的15号外显子存在一错义突变c.2282G>T(p.R761L);4b:与正常人pcr序列对比,患者母亲与患者同时出现15号外显子错义突变c.2282G>T(p.R761L)
患者2,男,33岁。手足背部及头面部、腹股沟丘疹26年来我院就诊。患者26年前双手出现丘疹,遇热后出现瘙痒,后头面部、腹股沟出现相似皮损,症状逐渐加重,曾于外院诊断“湿疹”,给予中药治疗,效果不佳,遂至我院就诊。家族史:其父亲及三个姐姐均有相似病史(图5)。体格检查:系统查体未见异常。皮肤科查体:手足背部多发对称分布的扁平肤色至褐色疣状丘疹,腋窝及腹股沟红棕色角化型丘疹,部分丘疹顶端破溃露出漏斗状凹陷。指(趾)甲未见异常(图6)。左手背部组织病理示:表皮角化过度,棘层轻度乳头瘤样增生,基底层局灶性棘松解。左腹股沟皮损示:表皮角化过度、角化不全,棘层增厚,基底层裂隙形成,基底层散在角化不良细胞,真皮浅层轻度淋巴样细胞浸润。病理诊断:毛囊角化病合并肢端角化症(图7)。基因测序发现,患者ATP2A2基因中15号外显子2300位发生了A→G错义突变(图8)。最终诊断:毛囊角化病合并疣状肢端角化症。治疗:给予口服阿维A胶囊10 mg日3次,后电话随访,患者自诉未规律服药,皮损变化不明显。

图5 患者2家系图
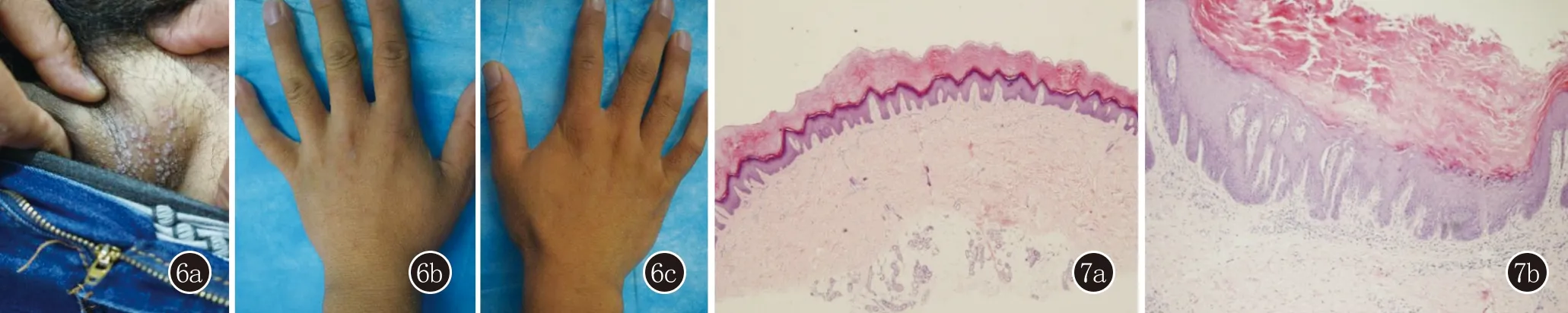
图6 6a:腹股沟部位坚实油腻淡灰红色丘疹;6b、6c:双手背部多发对称分布的密集肤色丘疹 图7 7a:左手背部表皮角化过度,棘层轻度乳头瘤样增生,基底层局灶性棘松解,呈疣状肢端角化症改变;7b:腹股沟部位表皮角化过度、角化不全,棘层增厚,基底层裂隙形成,基底层散在角化不良细胞,真皮浅层轻度淋巴样细胞浸润,呈毛囊角化病改变
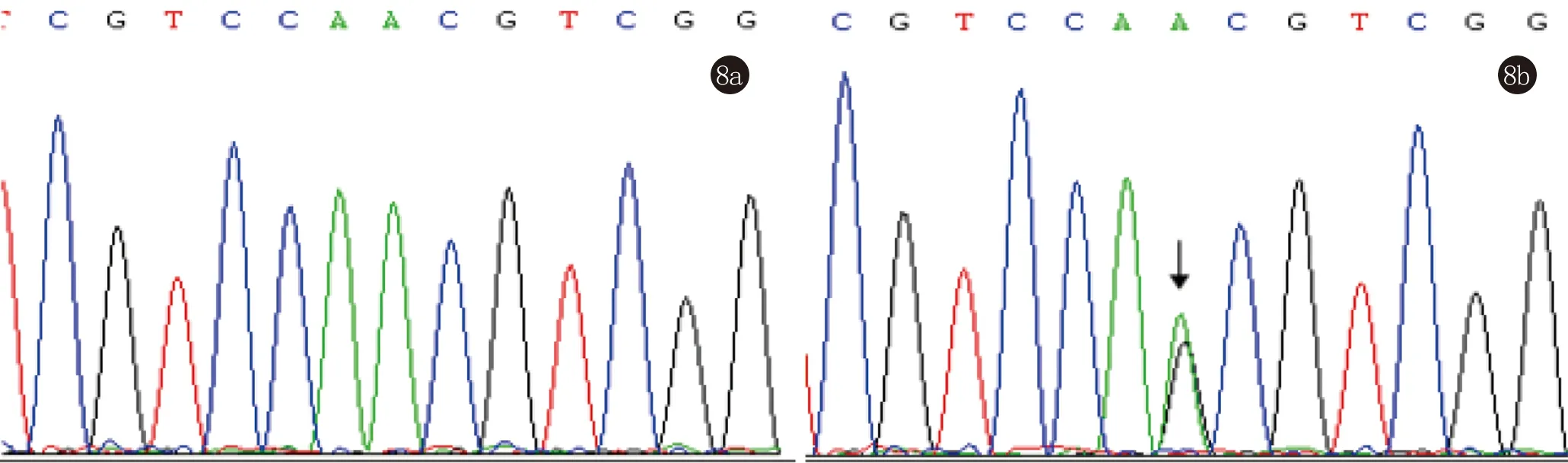
图8 与正常人pcr序列对比,患者2pcr结果示15号外显子错义突变c.2300A>G(p.N767S)
讨论AKV和DD都属于常染色体显性遗传病,都可以表现为手足的疣状丘疹。两者中都发现了ATP2A2基因的突变。关于AKV和DD是一种疾病的不同病谱还是两种不同的疾病,若干年来一直未定论。我们搜索了数据库“pubmed”近年来已被报道的疣状肢端角化症,对他们的临床表现等进行总结,并与毛囊角化病进行比较。我们共整理了30例AKV患者,收集了他们的发病年龄、家族史、临床表现、病理等特征(表1)。Burge等[1]对163例DD患者的临床特点进行归纳发现,68%的患者发病年龄在6~20岁之间,高峰发病年龄在11~15岁之间。在我们的总结中,AKV的平均发病年龄为16岁,与DD患病年龄相仿。67%的患者有家族史,皮损主要集中在手足和四肢。多达83%的患者累及足部,77%的患者有手部的皮疹,其中70%在手背,13%累及手掌。其余部位包括上肢37%,下肢37%,指甲27%。Burge的研究中,DD主要累及脂溢部位150例(92%)。角化丘疹发生在胸部(87%)和背部(85%),前额(84%)、锁骨上窝和颈部(82%)、头皮(69%)和耳朵(58%)。其中,有84%的人检测到掌部病变。46例(47%)患者无Darier病家族史。可以看出,AKV的皮损主要集中在手足和四肢,并不累及脂溢性区域。
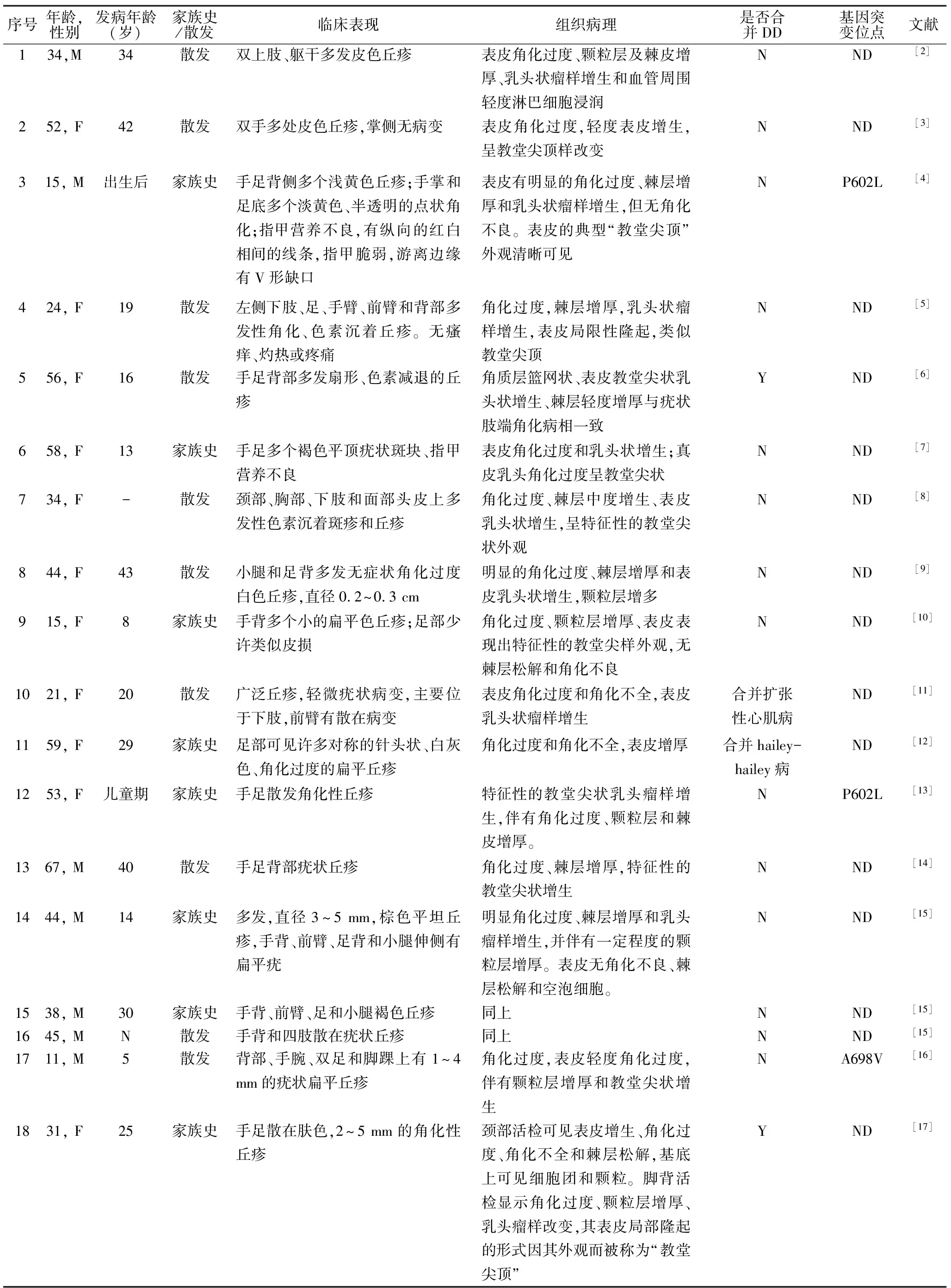
表1 30例AKV患者的临床特征
在本研究的30例患者中,4例合并DD,但均未进行基因检测。在行基因检测的3例AKV患者中,有2例检测到P602L 基因的突变,1例检测出A698V的突变。本研究中的2处突变位点均未在DD中报道过。在一个存在疣状肢端角化病的6代家系中被发现存在ATP2A2杂合子P602L突变后[25],我国学者张学军[26]调查了2个中国AKV家系和一例散发家系却未发现ATP2A2的突变,并提出可能存在其他基因的变异导致疾病的发生。
这些差异似乎倾向于AKV和DD是两种不同的疾病。然而,在涉及多个家系或三代以上家族的遗传调查中,AKV和DD共存的现象并不少见[27,28]。除此之外,一些学者还发现[29],在P602L突变家族的AKV患者的病理中发现了DD的组织病理特征。另一项关于成人DD和儿童DD差异性的研究[30]发现,在一组早期表现出AKV病变的患者中,除一例患者外,其他患者在成年后都发展出了DD的典型病变。根据这两种疾病间多种形式的重叠现象,有人推测AKV和DD可能是具有不同遗传表现形式的等位基因疾病[29]。
在我们的研究中,患者1的病理结果显示了AKV的特征。其突变位点与之前在DD患者中报道的相同。患者2两处部位的病理结果分别显示出AKV和DD的特征。经过基因检测,突变位点也符合之前报道过的DD突变位点。综上,我们推测两者可能是同一疾病在不同时期表现出的不同状态或因身体部位不同而表现出的不同病理特点。总之,AKV与DD之间的关系还需要继续探讨,通过提高AKV及DD基因检测率以及其他遗传学评估,并追踪这些患者的疾病发展情况可能有望解决这一难题。
