累及皮肤、眼、骨骼的Rosai-Dorfman病一例并文献复习
2022-08-20廖晓捷周桂芝王晓坤张福仁
廖晓捷 周桂芝 刘 红 王晓坤 张福仁
1山东第一医科大学附属皮肤病医院(山东省皮肤病医院),山东省皮肤病性病防治研究所,山东济南,250022;2山东第一医科大学附属山东省立医院,山东济南,250012
Rosai-Dorfman病(Rosai-Dorfman disease, RDD)是一种罕见的非朗格汉斯组织细胞增生性疾病,1965年被首次报道,1969年由Rosai和Dorfman命名为窦组织细胞增生伴巨大淋巴结病,特征表现为双侧无痛性、巨大的颈部或腹股沟淋巴结肿大,伴低热、体重减轻等全身症状[1]。皮肤Rosai-Dorfman(cutaneous Rosai-Dorfman disease, CRDD)是RDD的一种罕见类型,原发性CRDD继发眼眶受累并破坏骨质的病例尚未见报道,因此报道一例累及皮肤、眼和骨骼的RDD患者,并复习相关文献,以增加临床医师对该病的认识。
1 临床资料
患者,男,41岁。因右侧面部出现麻木感3个月、紫红色丘疹1个月,于2021年4月来我院就诊。患者3个月前无明显诱因右侧面部出现麻木感,后于相同部位出现紫红色丘疹1个月,伴轻度瘙痒,发病以来无发热、乏力、消瘦、关节痛等全身症状,既往史、个人史、家族史均无特殊。体检:系统检查未见异常,全身浅表淋巴结未触及肿大。皮肤科检查:右侧面部边界清楚的紫红色丘疹,粟粒大,表面光滑,皮肤略肿胀(图1a),皮温正常,无压痛,其余部位无皮损。实验室检查:血常规、肝肾功能、电解质、C反应蛋白、ANA、ds-DNA 均未见异常。皮肤浅表彩超显示:右侧面部皮肤层内探及多个小结节样低回声,无包膜,边界尚清,范围约0.35 cm×0.19 cm,内部回声尚均质,CDF1示内部未见血流信号。皮肤组织病理(图2):表皮轻度细胞间水肿,真皮内淋巴组织细胞、少许浆细胞、巨细胞、中性粒细胞浸润。免疫组化(图3):组织样细胞S100(+)、CD68(+)、CD1a(-)。诊断:皮肤Rosai-Dorfman病。治疗:口服沙利度胺50 mg/d,外用丁酸氢化可的松乳膏和0.1%他克莫司软膏每日1次,治疗2个月后皮疹完全消退。
7个月后患者自述面部原发皮疹处疼痛明显,于2021年12月再次于我院就诊,系统检查未见异常。皮肤科查体:皮疹完全消退,右侧面部皮肤略肿胀、质硬,下眼睑内眦处可扪及皮下肿物,边界不清,无波动感,有压痛(图1b)。皮肤浅表彩超显示:右侧面部皮下软组织内探及不规则实性低回声,范围约3.00 cm×2.91 cm×0.90 cm,临近骨质毛糙,回声增强,CDF1示可见点状血流信号。患者遂至外院眼科就诊,眼部CT平扫示右侧眼眶下壁见类似软组织密度灶,边界欠清,范围约2.5 cm×2.0 cm,包绕眶下壁,局部骨质破坏不连续,部分病灶向上突入眶内,向下突入上颌窦(图4)。诊断为右侧眶内异物,在全麻下行右眼眶肿物切除术,术中快速冰冻切片示纤维间质增生伴淋巴细胞及浆细胞浸润;术后组织病理示眶内增生胶原纤维组织中内见多量淋巴细胞、浆细胞浸润。免疫组化:浆细胞CD38(+)、Ki-67生发中心淋巴细胞(+),IgG(多量+)、IgG4(多量+),IgG4/IgG>40%。综合病程发展和术后病理,诊断为眼眶RDD伴骨骼受累,现术后随访2个月,未见复发迹象。
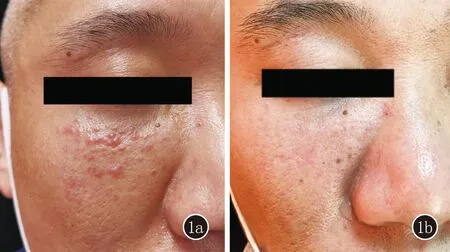
图1 1a:右侧面部紫红色丘疹,粟粒大,表面光滑,聚集成片,皮肤略肿胀;1b:治疗2个月后皮疹完全消退,7个月后右侧面部皮肤略肿胀、质硬

图2 表皮轻度细胞间水肿,真皮内淋巴组织细胞、少许浆细胞、巨细胞、中性粒细胞浸润(HE,×400) 图3 3a:组织样细胞S100染色阳性(IHC,×200);3b:CD68弱阳性(IHC,×100)
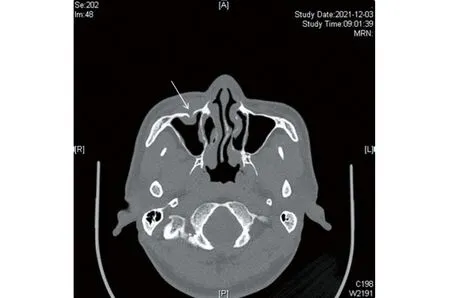
图4 眼部CT平扫示右侧眼眶下壁见类似软组织密度灶,边界欠清,范围约2.5 cm×2.0 cm,包绕眶下壁,局部骨质破坏不连续,部分病灶向上突入眶内
2 讨论
Rosai-Dorfman病是一种罕见的R组非朗格汉斯组织细胞增生性疾病,好发于儿童和年轻人,平均发病年龄为20.6岁[2]。该病可分为经典型、结外型、肿瘤相关型、IgG4相关型、免疫相关型和家族型。经典型RDD占19%,多累及淋巴结,表现为双侧淋巴结无痛性肿大,伴或不伴全身症状如发热、盗汗、体重减轻等,可伴结外多个器官受累,如皮肤及皮下组织、鼻窦、骨骼、乳房等[3]。
不累及淋巴结的结外型RDD约占43%,可涉及皮肤及皮下组织、呼吸道、骨骼和中枢神经系统等多个系统[4]。仅有皮肤受累的结外型RDD 称为皮肤Rosai-Dorfman 病(CRDD),仅占RDD患者的3%,但其在人口学特征、临床表现和预后上与RDD存在显著差异,被归为组织细胞增生的C组,其发病年龄较RDD晚,平均43.5岁,且在亚洲女性中更常见[5]。CRDD的皮损可单发或泛发,任何部位均可受累,以躯干部最常见,其次是头颈部。Al-Khateeb[6]总结发现CRDD的皮损多表现为无症状的黄色或红棕色簇状分布的丘疹或结节,也可融合成浸润性暗红色斑块,CRDD中仅11.2%累及面部,最常见于脸颊和眼眶区,多双侧分布。本例患者表现为面部紫红色的丘疹,粟粒大,但为单侧分布。
除皮肤外,结外型RDD可伴腮腺、眼睑、肝脏或乳房等的受累,但同时累及皮肤、眼、骨骼的RDD尚未见报道。本例患者在出现皮肤表现的同时,也出现眼及骨骼的受累。11%的RDD累及眼附属器,以儿童、青少年多见,表现为可触性肿物、眶周疼痛、眼睑下垂、眼球突出或运动受限、视力下降及压迫性视神经病变等,其中眼球突出是最常见的表现,受累部位以眼眶、眼睑和葡萄膜多见[7]。Al-Maghrabi等[8]总结发现眼眶RDD通常是单侧发病,无淋巴结受累的孤立性的眼眶病例极其罕见,且影像学无特异表现。本例患者中年男性,无眼球突变、视力下降等症状,仅表现为可触及肿物和眶周疼痛,应属于疾病早期,其眼眶CT表现为眶内占位,破坏骨组织和周围软组织,且突入上颌窦,影像学表现也无特异性。关于骨骼,10%的RDD可累及,骨骼RDD好发于青壮年,平均发病年龄为31岁,临床表现为局部的疼痛和肿胀,影像学主要表现为境界清楚、边缘硬化的溶骨性病变。Mosheimer等[9]总结分析,颅骨、颌面骨和胫骨是最常受累的骨骼,其中累及眼眶骨的RDD仅占0.08%。眼部RDD伴骨骼受累的病例更为罕见,Dahrouj等[10]于2019年首次报道RDD泪腺受累伴邻近的眼眶骨质破坏的病例。本例患者为皮下结节进展累及眼眶且伴邻近骨骼受累,仅表现为轻微的骨质连续性的中断,但影像学上也为溶骨性破坏。
RDD的诊断和鉴别诊断主要依靠病理学检查和免疫组化,组织病理具有典型的特征,高倍镜下可见深染区和浅染区,浅染区主要由体积较大、胞质丰富且核仁突出的组织细胞组成,组织细胞胞浆吞噬有完整的淋巴细胞和浆细胞,称为伸入运动,其对RDD具有重要的诊断意义但非特异性[11]。结外型RDD少见典型的伸入运动,而多为大量的组织细胞增生和慢性炎细胞浸润,易与IgG4-RD、郎格汉斯组织细胞增生症、结核、淋巴瘤等疾病混淆[12],所以确诊需结合病史及免疫组化排除其他疾病。免疫组化对该病的诊断至关重要,S100蛋白和CD 68阳性表达,CD1a不表达。本例患者的皮肤和眼附属器组织病理均未见典型的组织细胞伸入现象,且眼附属器组织病理的纤维组织增生明显,免疫组化虽有IgG4阳性浆细胞数目和IgG4/IgG比值的增加,但血清IgG4:0.755 g/L,未达到IgG4-RD的诊断标准。RDD可累及多个器官,确诊后建议行系统性的影像学检查排除淋巴结及其他器官受累。
RDD具有自限性,高达50%可自行缓解[11],无症状的患者主张随访观察,但对于有不适症状、皮疹泛发或进展累及其它器官的患者应给予治疗,包括手术切除、免疫抑制和免疫调节治疗等[13]。最有效的方法是手术切除,尤其适于皮损局限者;对难治性或多发皮损者,可系统应用或皮损内注射类固醇激素,但Goyal等[4]报道仅56%的患者经类固醇治疗有效,对疗效不佳者可与糖皮质激素联合应用或单用沙利度胺、氨苯砜、雷公藤和伊马替尼等免疫抑制调节剂,Liu等[14]报道1例应用来那度胺联合地塞米松治疗系统性RDD,治疗半年后,皮损和其它器官的软组织肿块均完全消退,说明联合方案可作为一线治疗。此外,也有次全切联合5-氨基酮戊酸光动力疗法、脉冲染料激光成功治疗CRDD的个例。
本病常为良性病程,预后较好,但部分有复发和进展累及多个系统的倾向。Liu等[14]报道1例RDD患者,其下肢皮损未经治疗进展延伸至上肢和躯干,7个月后患者出现鼻塞等不适症状,检查发现鼻窦、上颌窦、纵膈均受累;Karami等[15]报道1例面部CRDD的患者经手术切除治疗2年后,背部皮肤也出现受累,且皮下肿物复发于面部原皮损处并侵犯累及腮腺。本例患者中年男性,病程1年,皮损局限单一,但面部皮损经治疗消退后皮下结节进展累及眼眶破坏骨质,手术治疗后随访3个月暂无复发。因此针对原发于皮肤的RDD患者,病情有进展和复发的可能,且发生于面部的CRDD患者,注意面部器官如鼻窦、眼附属器、腮腺、泪腺累及的可能,应密切随访。
