EXT1基因突变导致遗传性多发性骨软骨瘤1例并文献复习
2022-06-21贾海亭李麒麟于嘉智王春华刘涛刘毅孙琳
贾海亭,李麒麟,于嘉智,王春华,刘涛,刘毅,孙琳
(1.山东大学齐鲁儿童医院a.骨科创伤外科,b.儿研所,济南 250022; 2.首都医科大学附属北京儿童医院骨科,北京 100045)
遗传性多发性骨软骨瘤(hereditary multiple exostoses,HME)是一种累及软骨化骨、以多发骨软骨瘤为特征的常染色体显性遗传病[1-2],1814年由Boyer首先提出[3],1964年Solomon[4]最先从基因角度研究该疾病。骨软骨瘤的形成主要由骺板处软骨细胞异常增殖突出相邻骨膜引起[5],软骨帽覆盖在其表面并充当骨骺生长板。根据肿瘤基底部形状,骨软骨瘤分为有蒂(有一窄茎,顶部较宽)及无蒂(基底宽而扁)两种[6-7],临床主要表现为干骺端骨性肿物,可引起疼痛、畸形、关节活动受限等。该病在欧美人群患病率约为1/50 000,由于存在无症状或症状轻微而未就诊患者,真实发病率可能会更高,目前国内尚无确切统计数据[8]。目前认为,EXT1、EXT2和EXT3基因与该病有关,其中EXT1和EXT2基因已被克隆,且大多数患者与这两个基因有关[2]。本研究报道1例由EXT1基因突变(c.1469delT)导致的HME患儿,其突变位点位于8号外显子,现对其临床特点、遗传学检测结果等进行总结,并通过文献复习HME的发病机制、临床表现、病理和影像学特点以及治疗方法等。
1 临床资料
患儿,男,2岁10个月,因发现全身多处无痛肿物1年于2020年9月在山东大学齐鲁儿童医院住院。现病史:入院前1年,家长无意中发现患儿双腕关节、双膝关节皮下有突起肿物,无红肿,质地硬,不伴发热等,肢体活动无受限,于山东大学齐鲁儿童医院门诊就诊,初步诊断为多发骨软骨瘤病,建议随诊观察。现患儿出现双上肢活动受限,为行诊治,遂于骨科创伤外科住院。既往史:既往体健,无输血史,无肝炎、结核等传染病史及接触史,无手术、外伤史,对头孢类药物过敏,无食物过敏史。个人史:系第1胎第1产,足月剖宫产,无窒息缺氧史,生后母乳喂养,5个月添加辅食。2个月抬头,6个月会坐,8个月会爬,1岁可自行走路,智力发育与同龄儿无差异。家族史:父母身体健康,非近亲结婚,患儿母亲的哥哥、父母、爷爷、奶奶无相应临床表现,否认家族遗传史。
体格检查:胸部形态正常,右侧胸部可触及骨性包块,质硬,双腕关节、双股骨远端内侧、右侧胫骨近端内侧可触及骨性包块,无红肿,质硬,左前臂旋转、旋后受限,左腕关节背伸功能受限,右腕关节旋前功能受限,左足第4足趾近节趾骨增粗。
辅助检查:实验室检查血常规、肝肾功能、凝血未见异常。双上肢、双下肢、胸部、骨盆等X线片显示双侧桡骨远端、右侧尺骨近端、右侧胫骨、双侧腓骨、双侧股骨远端见多处骨性突起,宽基底,背关节生长,骨皮质、骨小梁与母骨相连,未见钙化,周围骨质未见破坏及骨膜反应。左桡骨远端局部骨质密度减低,边缘呈条状高密度。左足第4趾近节趾骨增粗,局部可见骨性突起。右侧第5、7、8肋骨前端见骨性突起,骨皮质、骨小梁与母骨相连。双侧髂骨局部、双侧坐骨、耻骨及股骨近端局部边缘不光整,局部见丘状骨性突起(图1)。
治疗:因患儿左前臂功能受限较明显,遂全身麻醉下行左桡骨远端骨软骨瘤切除术。于左桡骨远端掌侧骨软骨瘤表面纵向切口,长约3 cm,依次切开皮肤、皮下脂肪、深筋膜,于肌间隙中暴露肿物,大小约3 cm×2 cm×1.5 cm,质硬,边界清,表面覆盖薄层软骨,将肿物包块软骨帽完整切除送病理。向远端尺侧暴露尺桡关节之间肿物,大小约2 cm×1.5 cm×1.5 cm,因掌侧切口不能完整暴露肿物,故切除肿物掌侧部分,取背侧尺桡关节纵切口,长约3 cm,依层切开暴露骨肿物,大小约1 cm×1 cm×1 cm,完整切除肿物,术中透视桡骨远端少量残留。术后病理示骨软骨瘤。术后左前臂功能改善,术后复查X线片见图2。

图1 遗传性多发性骨软骨瘤患儿双上肢、双下肢、胸部、骨盆X线片 1a、1b为左尺桡骨正侧位X线片,1c、1d为右尺桡骨正侧位X线片,1e、1f、1g为双侧胫腓骨正侧位X线片,1h为双足正位X线片,1i为胸部正位X线片,1j为骨盆正位X线片
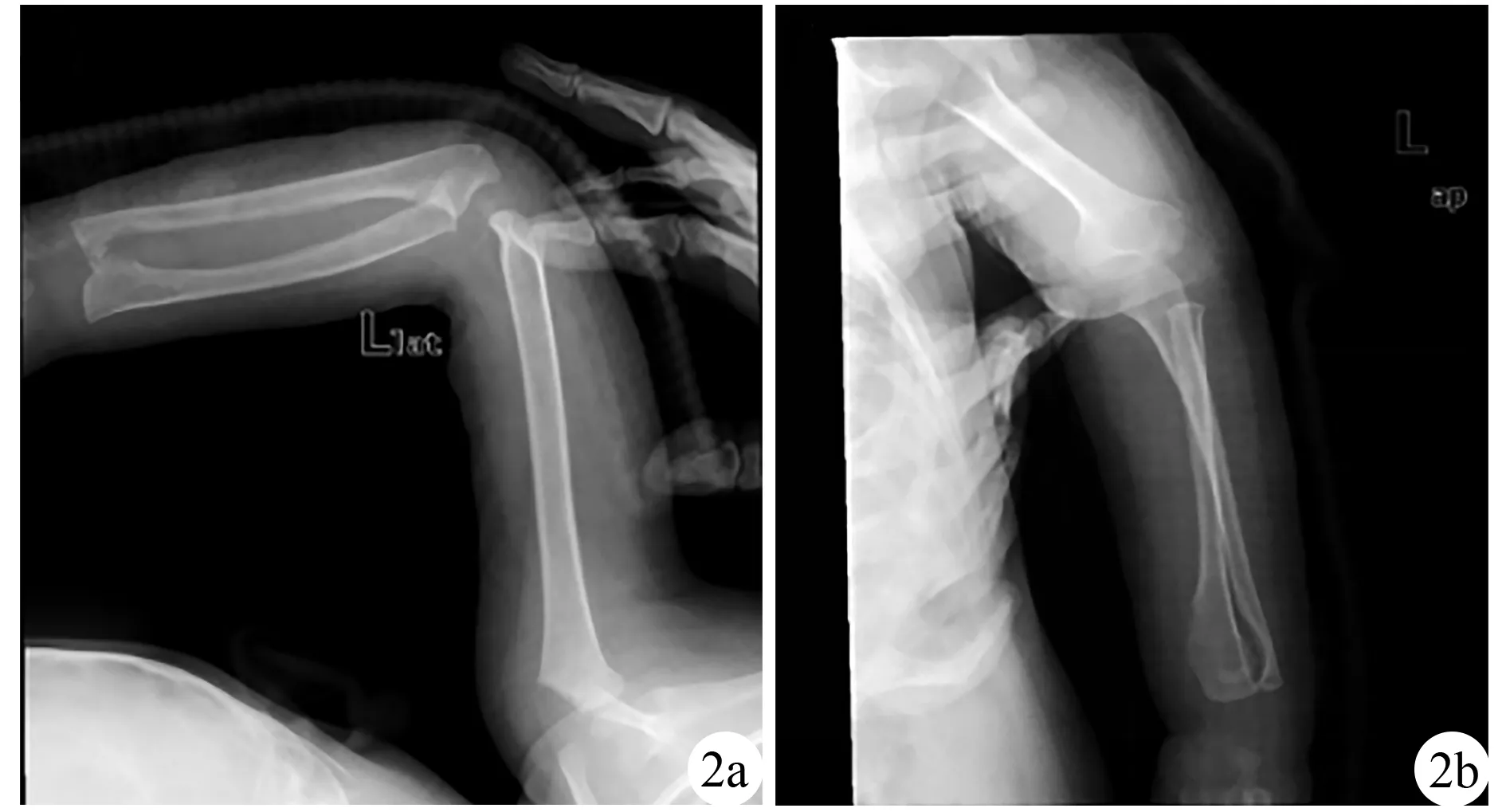
图2 遗传性多发性骨软骨瘤患儿术后复查左尺桡骨正侧位X线片 2a为左尺桡骨正位,2b为左尺桡骨侧位
基因检测:患儿家长签署知情同意书后,抽取患儿及其父母外周血,提取基因组DNA,构建基因组文库,然后通过探针杂交捕获与遗传性骨病相关基因的外显子及相邻内含子区域(50 bp),并进行富集。富集的目的基因片段通过一代高通量测序仪进行测序。对疑似致病变异进行Sanger测序验证。检测结果发现,患儿EXT1基因第8号外显子c.1469delT杂合突变,经Sanger测序验证发现该突变来自其母亲(图3)。对变异致病性进行分析发现,该变异导致其编码的蛋白从第490位开始翻译到第9位氨基酸时发生提前终止,预期对患儿正常功能有重要影响,且该变异为已报道的致病性变异。因此,该患儿最终诊断为EXT1基因变异导致的HME。
2 讨 论
HME又称骨干续连症、家族性外生骨疣、遗传性多发性外生骨疣、多发性软骨外生骨疣、遗传性畸形性软骨发育异常等,是一种以多发性外骨骼发育为特征的软骨生长紊乱性疾病,受累部位依次为长骨、肩胛骨、肋骨、脊柱[9],大多数患者有家族遗传史,临床表现主要为骨骼上大小不等的骨隆起,常呈双侧性和对称性,瘤体经常与骨骼的生长板相邻,且患儿生长发育结束前会一直生长。骨软骨瘤可导致许多健康问题,包括骨骼弯曲和畸形、活动受限以及神经和血管的压迫等[10-11]。据文献报道,约94%的患者有膝盖外生骨软骨瘤[12],其中约90%发生在股骨远端,84%发生在胫骨近端,76%发生在腓骨近端[6],约1/3 HME患者发生膝外翻[9]。前臂骨软骨瘤约37%发生在桡骨近端,38%发生在尺骨近端,80%发生在尺骨远端[12],在所有HME患者中有30%~60%存在前臂畸形[13],通常会引起肘部功能受限以及慢性疼痛[14]。肩胛骨骨软骨瘤占所有HME的3%~4.6%,多发生于肩胛骨腹侧[15],因腹侧面邻近胸廓,肿瘤压迫肋骨,与胸壁摩擦导致弹响和慢性疼痛,严重者可导致胸廓畸形、肺损害、肢体肿胀肌无力等[16-17]。肋骨骨软骨瘤约占所有HME的1%,多发生于肋骨前端肋软骨处,肿瘤伸入胸膜腔压迫肺部引起胸闷疼痛,严重者可导致自发性血气胸及冠状动脉压迫等[18]。脊柱骨软骨瘤临床较少见,发病率为1%~9%[19],其中30%~80%发生在颈椎,20%~30%发生在胸椎,腰椎较少见[20],由于椎管内空间狭小、结构复杂,且内容物性质特殊,肿瘤可压迫脊髓和神经根造成严重后果[21]。HME有一定的恶变倾向,恶变率约为2%,最高达25%,多恶变为软骨肉瘤或骨肉瘤。
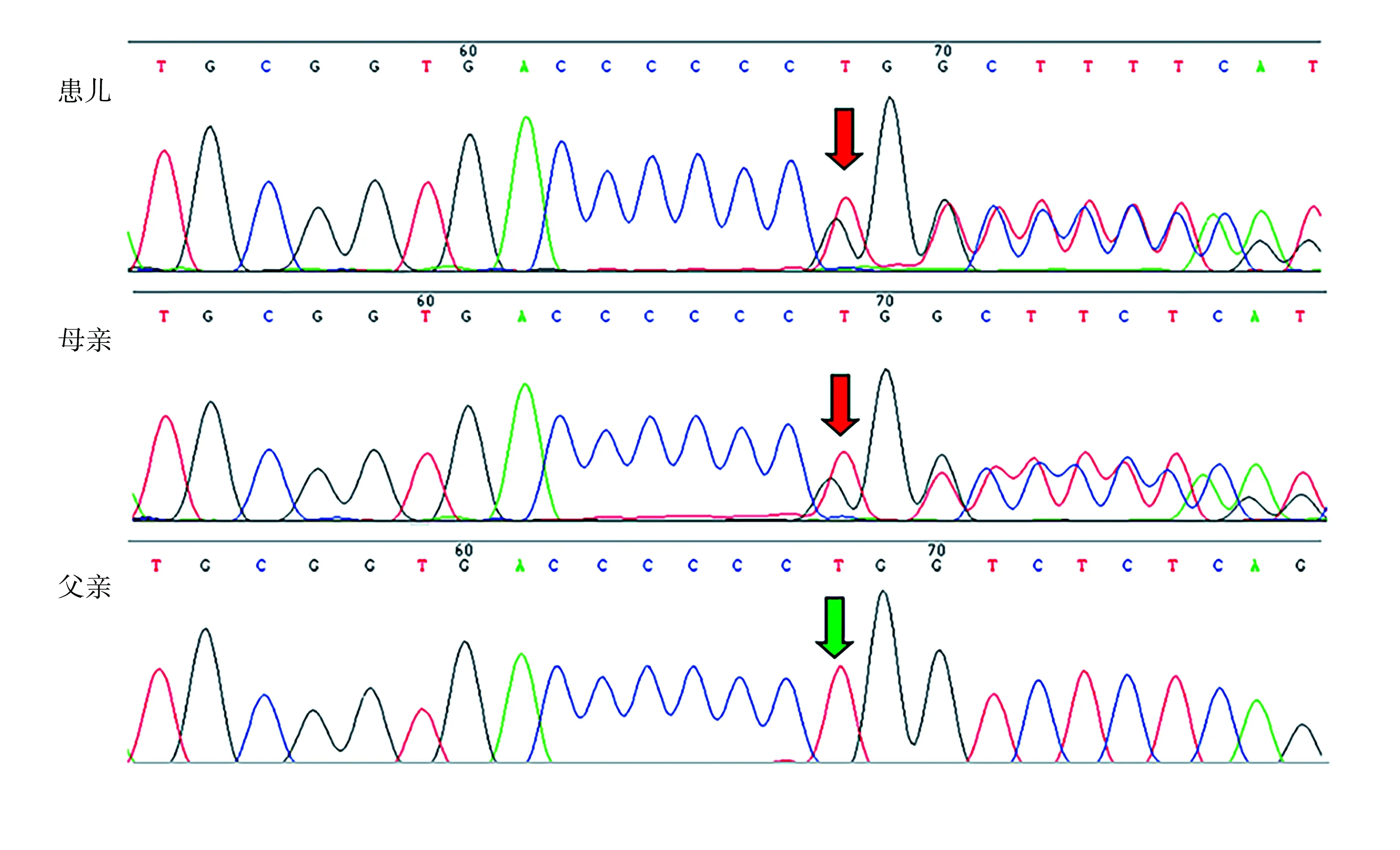
图3 Sanger测序的突变验证
HME发病原因主要与基因突变有关,约85%的HME病例被发现与EXT1或EXT2基因的杂合子功能缺失突变有关,15%的病例与EXT3、EXTL1、EXTL2和EXTL3有关[22],本例患儿基因检测EXT1基因c.1469delT杂合突变,为人类基因突变数据库报道的已知致病性变异。EXT1基因含11个外显子,编码2 238个位点,位于人类染色体8q24.11~q24.13。在HME患者中,10%的突变是自发的,80%的突变是截断突变,包括无意义、移码和剪接位点突变,这些突变通常在翻译过程中引入过早的终止密码子,或导致基因功能的部分或全部丧失[23]。EXT1基因编码的蛋白质为定位于内质网上的Ⅱ型跨膜糖蛋白,是具有催化作用的糖基转移酶,参与硫酸乙酰肝素的合成[24]。硫酸乙酰肝素是软骨的重要组成成分,广泛分布于体细胞周围和细胞膜,对体细胞生长和分化的调控以及其他多种细胞外基质蛋白的调节具有重要作用。EXT1基因发生突变导致硫酸乙酰肝素合成不足,从而引起软骨细胞过早分化,使邻近区域的骨骼异常生长,进而引起多发性外生性骨软骨瘤。然而有文献报道,硫酸乙酰肝素的缺乏不一定会导致骨软骨瘤的形成,当硫酸乙酰肝素的水平显著降低但未完全消失时,如杂合性丢失或复合杂合突变等二次因素可能是导致疾病发展的原因[9,25]。
本例患儿母亲携带EXT1基因的变异位点,但无临床表型,与文献中该基因变异女性携带者外显不全的报道相符[26-27]。在EXT家系中,一部分女性携带突变基因但无临床表型,而男性的外显率更高、症状更严重;此外,轻型患者中女性比例更高,而男性重型患者更多见,关于上述差异有学者认为,一方面性激素在疾病进展中起到重要作用,其中雌激素在骨软骨瘤发生发展中有保护作用,而雄激素起有害作用;另一方面存在某些未知的X连锁基因对骨软骨瘤发病有影响,所以女性外显率较低,病情进展也相对较轻[25]。
HME的X线片表现为骨皮质向外骨性隆起,表面覆盖软骨帽,顶端光滑,通常有规则的骨髓内部结构。CT可直观立体多角度显示边界清楚的骨软骨瘤及其表面软骨帽,在评估HME相关并发症(脊髓压迫、血气胸、假性动脉瘤、骨质、肌肉神经组织受压等)中具有重要作用[28]。磁共振成像(magnetic resonance imaging,MRI)表现为皮质骨在T1加权成像上呈低到中信号,松质骨在T2加权成像上呈高信号,软骨帽在T1加权成像上呈低信号,在T2加权成像上呈稍高信号,钙化部分均呈低信号[2]。软骨帽在B超上呈低回声,B超能够评估软骨帽的厚度[29],同时能够检测出动静脉血栓、动脉瘤等血管并发症[30],但B超较多地依赖于操作者的技术水平,对位于软组织深部的软骨帽往往探查不清。一般通过X线即能诊断HME,但在骨盆、脊柱、胸骨等复杂解剖结构部位MRI和CT具有重要价值[31]。MRI能够较好地评估骨软骨瘤引起的滑囊炎、肌肉压迫、关节退行性病变等软组织改变;此外,Jurik等[32]通过随访观察发现,MRI在筛查HME恶变方面具有潜在的临床意义。
HME病理组织学特点类似单发性骨软骨瘤,在光镜下可见特征性的3层结构,最外层为薄层纤维组织,覆盖在珍珠白色软骨帽表面,软骨帽厚度不一,软骨组织内有大量呈聚集分布的增生瘤样细胞,细胞核较大,细胞质内可见圆形或椭圆形的线粒体及扩张的粗面内质网,软骨细胞体积较正常细胞增大,软骨细胞无或轻微典型性,无活跃的有丝分裂。软骨帽底部可见软骨骨化并与松质骨样区域融合,软骨基底新形成的骨小梁类似正常生长板的松质骨。如发现软骨细胞增多、胞体肥大、核大深染以及双核瘤细胞或多核瘤巨细胞和病理性核分裂象,则提示肿瘤发生恶变[33]。王永堂[34]通过扫描电镜和透射电镜观察儿童HME软骨帽的超微结构,发现儿童瘤体软骨帽软骨组织代谢旺盛,细胞增生及蛋白质合成活动活跃,部分瘤体软骨帽胶原纤维致密有钙化倾向,具有使瘤体恶变的一定物质基础,表明软骨帽是瘤体恶变、复发的根源所在。
对于大多数HME患儿,一般无须手术治疗,可每1~2年骨科门诊进行临床体格检查和X线检查,但当出现肢体畸形、疼痛、神经血管受压、功能活动受限以及恶变时往往需要手术切除或矫形。手术切除可以解除瘤体组织占位,减去对邻近组织的压迫,能够增加关节活动度、改善外观,延缓或阻止骨软骨瘤的进一步生长[35]。手术时要完整切除软骨帽,同时也要注意骺板的保护,如果彻底切除瘤体损伤骺板,则会影响骨骼的正常发育,但如果因为保护骺板瘤体切除不彻底,则会出现病变的复发,甚至可能因为手术的刺激引起病变恶化。对于手术年龄的选择观点不一,有学者认为患儿年龄越小,骨骼塑形能力越强,肢体畸形矫正效果越好[23];也有学者认为如无明显肢体畸形或功能活动受限,最好不要在儿童期手术,因为骨骼未成熟前行手术治疗复发率较高[36]。本例患儿发病年龄2岁10个月,但已有左前臂功能受限表现,因此行左桡骨远端骨软骨瘤切除术,但因术中保护骺板,因此桡骨远端有少量残留。目前在具体手术方式上尚未达成共识,对于前臂骨软骨瘤常用的手术方式有单纯肿瘤切除、尺骨延长、桡骨短缩等,Ilizarov外固定架通过缓慢稳定的牵拉可以激活骨骼代谢诱导骨骼再生[37],在HME前臂畸形的矫治中得到广泛应用。
对于大多数HME患儿,往往因肿物进行性增大或关节活动受限首诊于骨科,作为小儿骨科医师,焦点也主要集中在切除肿物改善功能,患儿家长和医师往往对基因检查不够重视和了解。本例患儿全身多发骨肿物,诊断为HME,但其父母均无临床表型,也没有家族史,因此对于“遗传”比较困惑,通过基因检测解答了该患儿致病基因来源于其母亲,同时基因结果可用于遗传咨询。因此在今后临床工作中,小儿骨科医师应重视基因检查,一方面可以明确病因,另一方面可用于遗传咨询。但本研究仍存在一定不足之处:①本例患儿因保护骺板导致肿物残留,其远期效果及前臂功能需要长期随访观察;②对于软骨瘤切除时机以及软骨瘤导致的肢体畸形最佳矫治时机需要进一步研究和总结。
综上所述,HME是一种累及人体骨骼系统影响正常软骨化骨的遗传性疾病,诊断主要依靠临床特征和基因检测,当出现肢体畸形、功能活动受限时需要手术切除。本研究中EXT1基因c.1469delT杂合突变是导致该患儿发病的分子机制,其母亲有外显不全,研究结果可用于遗传咨询。
