酱香型酒醅堆积发酵过程中优势菌群和稀有菌群对细菌总群落演替模式的影响
2022-02-16吴福勇李芳香钟艳霞莫新良王新叶
赵 亮,闵 卓,吴福勇,江 璐,李芳香,钟艳霞,张 阳,莫新良,何 惠,王新叶
(茅台学院酿酒工程系,贵州 遵义 564507)
堆积发酵是酱香白酒酿造的独特关键工序,属于边糖化、边发酵方式。高温大曲的曲药微生物能在酒醅上增殖、发酵,同时高温堆积网罗、富集环境空气中的微生物,生成酱香或酱香前体物质,为入窖发酵创造条件,相当于“二次制曲”[1-5],也为入窖发酵提供糖化基础和微生物储备[6-7]。
酱香型白酒中最重要且研究最多的微生物类群是细菌,其主要来自于高温制曲和堆积工序。细菌及其代谢产物在高温操作过程中强化了糟醅自身形成酱香的原动力,促进酱香物质的进一步生成[8]。因此,堆积发酵的细菌菌群结构及其变化对后期窖内产酒和生香具有举足轻重的作用,而研究酱香型白酒发酵过程细菌菌群结构是解析发酵机理和风味形成的基础保障[5],在近几年引起众多学者的关注。程才璎等[9]使用变性梯度凝胶电泳技术分析了酱香型白酒7个轮次细菌分布规律,发现同一轮次堆积发酵的前期、后期和中期多样性依此增大,Bacillus、Thermoactinomycetaceae、Staphylococcus、Virgibacillus、Lactobacillus、Corynebacterium、Streptomyces、Lentibacillus及一些不可培养类群为优势细菌群。Wang Li等[10]使用高通量测序(high-throughput sequencing,HTS)技术,发现Bacillaceae和Lactobacillales分别为堆积前期和窖内优势细菌群,并且多数来自大曲的细菌对堆积发酵前期的群落结构形成有重要作用。戴奕杰等[8]使用HTS技术分析酱香型酒醅整个生产周期所有轮次细菌群落结构,结果显示在产酒发酵阶段Escherichia-Shigella、Lactobacillus、Clostridium_sensu_stricto_1、Streptococcus、Actinobacillus、Bifidobacterium和Alternaria等细菌属为优势菌群。胡小霞等[5]用HTS技术分析酱香型酒醅1轮次发酵过程群落分布,结果表明在门和属水平上,堆积发酵检出的总细菌、主要细菌和优势细菌种类均比窖池发酵同比数据多,堆积发酵过程优势细菌属有Lactobacillus等13个,其中Lactobacillus、Escherichia-Shigella和Bacillus占主导地位。黄蕴利等[11]使用HTS技术针对酱香型酒醅第2轮次发酵过程微生物演替分布作分析,发现堆积酒醅中优势细菌有Bacillus、Enterococcus、Lactococcus、Lactobacillus、Lentibacillus、Kroppenstedtia、Enterobacteriaceae、Alkaliphilus、Oceanobacillus、Thermoactinomyces10 种。此外,众多同类型研究也对酱香型酒醅堆积发酵过程微生物多样性及群落结构作了详细描述,在此不一一列举。
在HTS技术成本廉价的近几年,针对白酒酿造过程微生物群落结构及多样性的研究如雨后春笋般盛行,其成果也加速了对白酒酿造微生物在群落水平的认识。研究焦点基本落于优势菌群(abundant taxa,AT),即通常单样本中相对丰度≥1%或所有样本中平均相对丰度≥0.5%的可操作分类单元(operational taxonomic units,OTU)组成的微生物子群。而在巨大的HTS数据集中,数量占比超过总OTU数50%、总相对丰度通常低于10%的稀有菌群(rare taxa,RT)往往被忽视。RT因丰度极低、数量巨多,也被定义为OTU秩-丰度曲线中的“长尾”菌群[12-13]。RT虽然丰度很低,但它们仍作为一个巨大的遗传与功能多样性储备库,对生态系统的功能和稳定性起潜在调节作用[14-15]。随着近些年对RT的逐步重视,已有其他领域的相关研究证明了RT的生态重要性。如RT比AT有更高代谢活性[16-17];同时,RT在功能上不同于AT,进而为总群落(whole taxa,WT)提供特定的代谢通路或补偿性功能,促使整个群落功能更加完善[18]。此外,AT和RT对环境的不同响应,也导致这两类子群在维持生态系统稳定性上的功能不同[17,19-20]。综上,先前研究已在其他微生物环境领域展现出各类子群落对WT的不同作用和影响,但目前基于此方向针对发酵体系的研究仍鲜有报道。酱香型白酒第1、2轮次的发酵分别对应生产上的下沙和造沙轮次的发酵,在这2个阶段需要加入新粮,为原粮发酵和微生物储备阶段。从第3轮次发酵开始不投入新粮,正式进入酒醅发酵阶段[10,21]。此阶段产出的2轮次基酒能够增加基酒中酒体的曲香味,使糟醅中累积更多的微生物代谢产物,有利于基酒香味物质的积累,在后期轮次相应形成更多的酱香型酒前驱物质,同时为后期轮次多产酱香奠定基础。因而第3轮次的发酵是后续发酵轮次的基础,也是后续各轮次酒的质量保障[11]。基于此,本研究以酱香型酒醅第3轮发酵轮次堆积过程为代表,分析AT和RT的演替及其驱动力,阐释两种子群对WT演替的推动作用,以及它们各自维持WT稳定性的功能角色。以此为进一步理解发酵体系微生物群落功能及生态稳定性提供理论基础及参考思路。
1 材料与方法
1.1 材料与试剂
1.1.1 样品
样品取自贵州茅台镇一酱香酒厂。堆子上取样深度及部位如图1所示,从上到下每个层面对应的深度a~e选取温度相同的5个点取样,均匀混合后作为对应层面的样品。如位于顶层选取深度30 cm, 温度相同的5个点取样,均匀混合后作为顶层的样品,依此获得顶层至底层的酒醅样品。按此取样方法,从丢堆第1天(收堆完成)至第8天(堆积结束),分别得到第1天(D1,0 h)、第3天(D3,49 h)、第5天(D5,95 h)、第7天(D7,143 h)和第8天(D8,164 h)共25个酒醅样品。取样时车间温度9~14 ℃,相对湿度55%~65%。
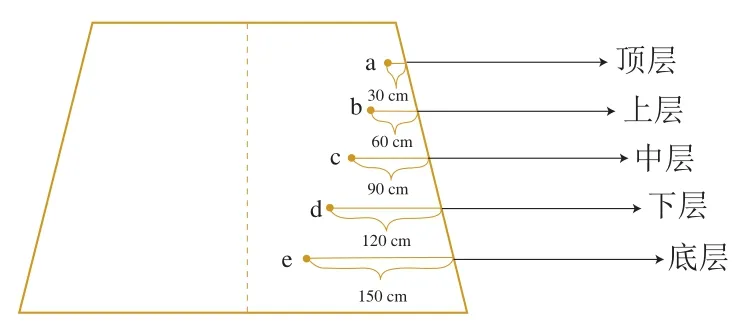
图1 堆子不同部位取样示意图Fig. 1 Sample collection from different positions in the stack
1.1.2 试剂
DNA提取试剂盒MP FastDNA®SPIN kit for soil美国MP Biochemicals公司;磷酸盐缓冲液(phosphate buffer solution,PBS)、TBE(Tris-硼酸-EDTA)缓冲液、异丙醇(分析纯)、琼脂糖 北京索莱宝科技有限公司;引物合成 杭州联川生物科技有限公司。
1.2 仪器与设备
台式高速冷冻离心机 德国Sigma公司;Gene Amp®9700型聚合酶链式反应(polymerase chain reaction,PCR)仪美国ABI公司;DYY-8C型电泳仪 北京六一仪器厂;SW-CJ-1F型超净工作台 苏州净化设备有限公司;HH-6数显恒温水浴锅 国华电器有限公司;Illumina MiSeq PE300测序平台 杭州联川生物科技有限公司。
1.3 方法
1.3.1 样品预处理及DNA提取
参照戴奕杰等[8]方法对酒醅样品进行预处理。预处理后得到的细胞沉淀,使用DNA提取试剂盒MP FastDNA®SPIN kit for soil,按说明书提示,提取酒醅样品中微生物细胞总DNA。
1.3.2 细菌16S r RNA基因扩增
使用引物319F(5’-ACTCCTACGGG AGGCAGCAG-3’)和806R(5’-GGACTACHVG GGTWTCTAAT-3’)[22]对细菌16S r RNA基因V3-V4可变区进行扩增。扩增程序:98 ℃预变性30 s,35个循环(98 ℃变性10 s,54 ℃退火30 s,72 ℃延伸45 s),72 ℃终延伸10 min。凝胶电泳:2%琼脂糖凝胶,核酸染料(GenGreen),电压90 V,电泳时间30 min。
1.3.3 Illumina MiSeq测序
采用Illumina HTS技术,基于Illumina MiSeq PE300测序平台,由杭州联川生物科技有限公司完成测序。
1.3.4 酒醅内源性理化指标检测
微生物在堆积过程经生长代谢,产生的生物热会积累在堆积酒醅当中,并作为内源因素影响微生物生长,堆子内的水分也是影响微生物生长繁殖的重要因素,因此堆子的水分含量和温度常作为酒醅发酵的重要表征参数。堆子内部有大量酵母菌,氧气不足时会产生乙醇,从而影响其他微生物生长[23],因此乙醇也是堆积过程的重要内源指标。乳酸和乙酸是白酒中重要的有机酸,同时也直接反映酒醅在发酵过程中是否正常。特别是乳酸,它是最主要调节酱香型白酒酒醅中pH值环境的物质,乳酸的积累与酒醅pH值环境密切相关,直接影响微生物群落的演变和发酵进程。另外,乳酸又是不同类型白酒中总酸的主要组成部分,最高含量可达90%以上。因此,酱酒生产中当需要探索酸环境与微生物之间影响关系时,通常乳酸最具表征性且作为主要内源指标[24-26]。综上所述,本研究选取温度、乙酸、乙醇、乳酸和水分5 种表征酒醅理化状况内源性指标。温度在取样时使用手持式温度计测量并记录。水分含量使用差重法,称取10 g新鲜酒醅样品于135 ℃烘干1 h后称质量,并计算水分质量分数。参考Song Zhewei等[27]方法使用气相色谱-质谱联用仪测定乙酸和乙醇含量。参考Bai Dongmei等[28]方法使用反相高效液相色谱测定乳酸含量。
1.3.5 高通量数据处理
PE300双端测序reads使用FLASH拼接,根据Barcode区分序列样本来源,使用QIIME分析管道[24]完成去偶联、质控、解析等过程。原始reads中含有超过3个连续碱基、质量评分小于Q20等级以及在同源匹配区域大于6个模糊碱基的序列一律被过滤掉;此外,使用USEARCH识别并除去嵌合体序列,以此获得可供分析的高质量有效序列[29]。使用UCLUST在97%序列相似度水平获得OTU[30],并从每个OTU中选取质量最好的序列作为对应OTU的代表序列。每个OTU代表序列利用RDP Classifier生成对应OTU的系统分类信息[31-32]。生成的OTU_table文件除去总序列数小于5的低丰度OTU,并利用R软件对OTU序列数进行重抽样,保证每个样品的总序列数完全一致,最终获得用于统计分析OTU_table文件。
1.4 统计分析
所有分析使用R软件各类程序包完成。
参照已有研究[19,33-34],并结合本研究数据结构特点,按以下方式将WT即所有OTU分成3个子群。AT:在所有样品中平均相对丰度≥0.05%,并且至少在1个样品中相对丰度≥1%。RT:在所有样品中平均相对丰度<0.001%,并且在任一样品中相对丰度<0.1%。条件稀有菌群(conditionally rare taxa,CRT):根据Shade等[35]报道,在群落中通常丰度很低,属于RT的一种,但在特定条件下丰度会瞬间变高,这一类RT称为CRT。本研究以每个OTU在D1~D9各时段平均相对丰度为数据基础,利用Shade等[35]双峰分布模型,计算每个OTU的双峰状态系数b,即:
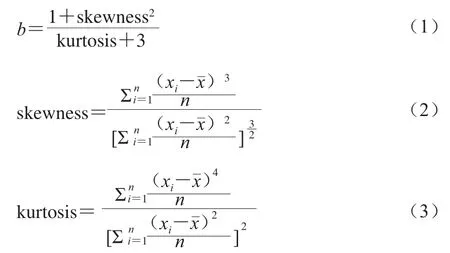
式中:xi为第i个OTU的相对丰度;为第i个OTU平均相对丰度。
凡双峰状态系数b>0.9、最高相对丰度>0.1%的OTU可被认定为CRT。
α多样性使用Shannon指数和Chao1指数衡量,β多样性使用Bray-Curtis非相似度矩阵衡量。群落演替分别使用物种-时间关系(species-time relationship,STR)模型[36-37]和时间-衰减关系模型(time-decay relationship,TDR)[33]衡量。参考White等[38]移窗法将时间分成多个区间,基于物种丰富度在不同时间区间的积累变化量,拟合STR模型[36-37],即S=k×Tz,其中S为物种丰富度Chao1指数,k为估计参数,T为时间间隔,z为物种积累变化率。TDR基于最小二乘线性回归Ss = constant-w×T,估计群落相似度随时间推移的变化趋势[39]。其中Ss为群落相似度(1-Bray-Curtis非相似度),T为时间跨度,constant为模型估计参数,w为群落相似度随时间推移的变化速率,即群落结构演替速率。
相似度检验和非参数多元方差分析检验样本分组间(时间段间)群落结构差异性;Mantel检验分析子群落的群落结构差异对WT(基于所有OTU)的群落结构差异的影响;使用限制对应分析(constrained correspondence analysis,CCA)检验影响各子群落演替的内源理化因子。基于Spearman相关性,分析微生物间共存网络。Spearman相关性系数r≥0.6,显著性P值经错误发现率(false discovery rate,FDR)矫正后的显著性值q小于0.05的OTU用于构建网络,并使用Gephi软件呈现网络图[40]。分别在节点水平和子网络水平计算各子群在网络中的拓扑系数[41-42]。
2 结果与分析
2.1 α多样性及群落结构分布
原始序列经质控、OTU聚类和样本reads数均一化处理,共得到493 050 条reads,6 943个OTU,每个样本包含19 722 条reads。将这些OTU按AT、RT和CRT分成3个子群,每个子群及WT的α多样性信息如表1所示,AT丰度占比为84.1%,RT为1.8%,CRT为12.0%;AT的OTU数量占比为0.4%,RT为70.9%,CRT为0.1%。Shannon指数和Simpson指数显示RT多样性显著高于其他子群及WT;Pielous指数显示RT均匀度也显著高于其他子群及WT。这个结果表明,RT可能是维持发酵系统群落稳定的重要保障。因为在生态系统中,微生物体系的多样性是维持生态系统功能稳定性的重要条件[43-45]。
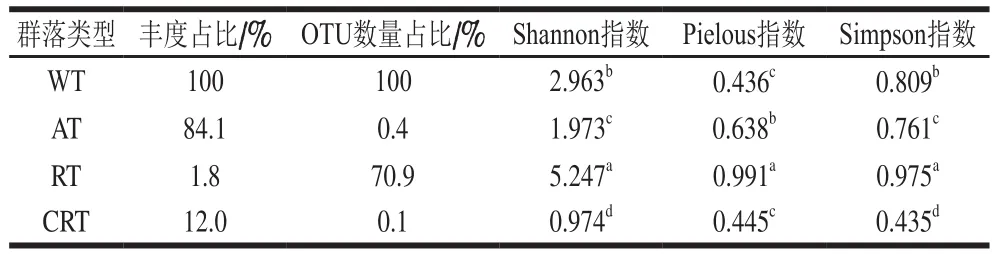
表1 细菌WT和各子群α多样性信息Table 1 Information about alpha diversity in whole bacterial community and each sub-community
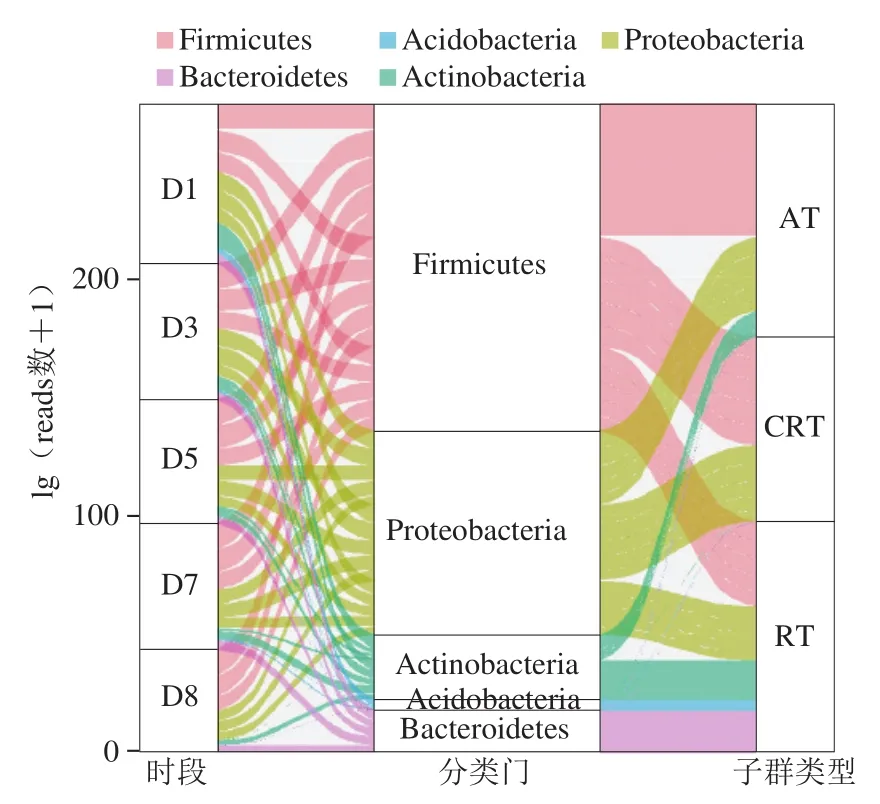
图2 发酵过程各子群在门水平群落结构分布图Fig. 2 Alluvial diagram showing the distribution of sub-community composition at the phylum level during stacking fermentation

图3 AT、RT和CRT丰度时空分布图Fig. 3 Temporal and spatial distribution of mean abundances of the three sub-communties
3个子群在门水平的分布显示(图2),AT仅由厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria)组成;RT主要由厚壁菌门、变形菌门、放线菌门、酸杆菌门(Acidobacteria)和拟杆菌门(Bacteroidetes)组成;CRT仅由厚壁菌门和变形菌门组成。属水平,AT由芽孢杆菌属(Bacillus)、慢生芽孢杆菌属(Lentibacillus)、嗜热放线菌属(Thermoactinomyces)、乳酸杆菌属(Lactobacillus)、火山渣芽孢杆菌属(Scopulibacillus)、Kroppenstedtia、海洋芽孢杆菌属(Oceanobacillus)、欧文氏菌属(Erwinia)、片球菌属(Pediococcus)和甲基杆菌属(Methylobacterium)组成。RT除了包含上述各属外(不含片球菌属),还主要由假单胞菌属(Pseudomonas)、肠杆菌属(Enterobacter)、魏斯氏菌属(Weissella)、泛菌属(Pantoea)、葡萄球菌属(Staphylococcus)、肠球菌属(Enterococcus)、梭状杆菌属(Clostridium)、乳酸乳球菌属(Lactococcus)、异常球菌属(Deinococcus)、明串珠菌属(Leuconostoc)、枝芽孢菌属(Virgibacillus)和不动杆菌属(Acinetobacter)组成。CRT由嗜热放线菌属(Thermoactinomyces)、芽孢杆菌属(Bacillus)、欧文氏菌属(Erwinia)、乳酸杆菌属(Lactobacillus)、肠杆菌属(Enterobacter)和肠球菌属(Enterococcus)组成。这些微生物虽然在先前研究中有所报道,但本研究阐明了这些微生物具体的子群落归属,对深入理解发酵过程微生物区系的群落结构提供分析依据。此外,从3 类子群在时空上的平均丰度(平均reads数)变化趋势可以看出(图3),AT和CRT较为相似。时间尺度上,AT和CRT平均丰度在D1时最低,分别为528和195;D3~D7相对较高,AT平均丰度为696~678,CRT平均丰度为277~288;至D8时有所回落,AT和CRT平均丰度分别为630和240。空间尺度上,AT和CRT在堆子上层(AT:596;CRT:236)和下层(AT:599;CRT:210)平均丰度相对较高,而在顶层(AT:528;CRT:195)、中层(AT:581;CRT:186)和底层(AT:522;CRT:188)相对较低。RT平均丰度的变化趋势与AT和CRT相比略有差异。时间尺度上,RT在D1~D7呈下降趋势(0.17~0.05),至D8(0.09)稍有回升。空间尺度上,RT在下层(0.19)和底层(0.19)相对丰度较高,其次为顶层(0.17)和中层(0.16),上层(0.14)最低。这些结果初步表明,RT与AT和CRT相比,具有相对独立的演替模式。
2.2 β多样性分析
Mantel检验结果显示,WTβ多样性分别与AT、RT和CRT的β多样性显著相关(P<0.01),说明3个子群落的结构演替对WT的结构演替都有贡献。从相关性系数r看,AT最高(r=0.985),其次是CRT(r=0.908)和RT(r=0.793),表明AT是WT结构演替的主体,代表WT演替的方向和趋势,CRT对WT演替起到一定推动作用,RT与AT和CRT相比,在WT中的演替相对独立。此外,3个子群落在WT中β多样性占比也可看出,AT是推动WT演替的主体群落,占比31.5%;其次是CRT,占比4.5%;RT对WT演替影响最小,占比1.8%。WT和3个子群分别拟合STR模型发现,仅WT和RT存在极显著z值(P<0.001),分别为-0.48和-0.68,其他两个子群的z值无显著性(AT:z=0.005;CRT:z=0.02;P>0.05)。说明发酵过程中WT的物种积累数以0.48的速率呈指数趋势缩减,且来自于RT的快速缩减。此外,WT和3个子群拟合TDR均可得到极显著的负斜率(P<0.001,图4),即随发酵时间的推移,酒醅内群落结构会以恒定的速率发生演替。3个子群落中,CRT陡度最高(|w|=0.047),演替速率最快,AT(|w|=0.034)次之,RT演替速率最慢(|w|=0.008)。结合WT演替速率(|w|=0.037),可以看出,CRT和AT是决定WT演替速率的主要菌群。Carvalho等[46]认为群落β多样性,即群落结构演替变化由物种丰富度和物种丰度两方面因素决定。使用STR和TDR两种模型可分别从物种丰富度和物种丰度2个角度探究微生物群落在发酵过程中如何演替以及演替的内驱菌群,并且对微生物演替的速度作定量描述。先前关于酿酒微生物群落结构演替的研究,主要聚焦于微生物系统分类群如何随时间或空间的变化而发生改变。本研究从群落演替的动力角度入手,为深入剖析发酵过程群落结构演替规律,并通过演替速率间接认识发酵动力提供理论参考。
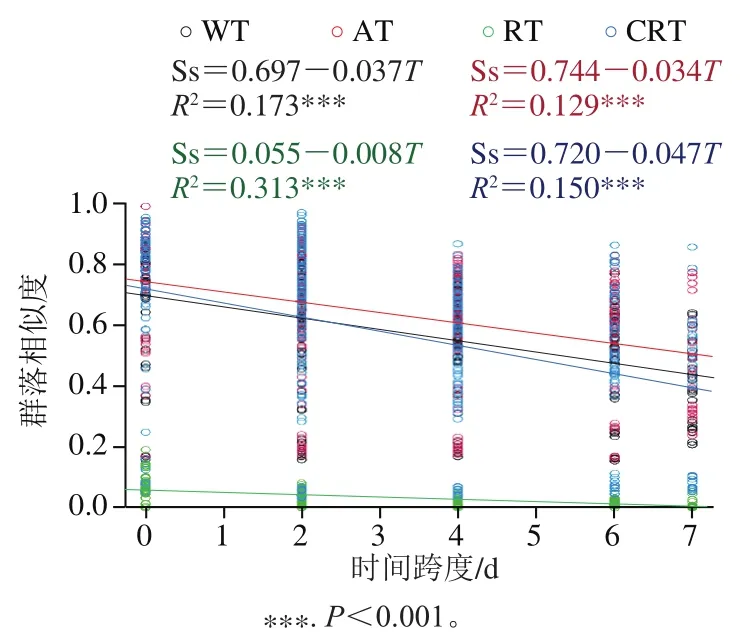
图4 群落相似度与时间跨度关系曲线图Fig. 4 Curves showing community similarity against time span
2.3 相关性网络分析
利用Spearman相关性构建共存网络(图5),网络内共有6 898个相互间显著相关(q<0.01)节点(OTU),816 922个连接边。其中,AT节点24个,CRT节点4个,RT节点4 914个,其他节点1 956个。分别对AT、CRT、RT以及不属于这3种子群类型的其他子群(others)计算节点水平网络拓扑系数(表2),可以看出RT中介中心度、接近中心度和平均连接度均高于其他子群,且接近中心度和平均连接度显著高于其他子群。此外,RT的平均最短路径长度也显著短于其他子群。此结果表明RT相比其他子群更加处于整个网络的中心位置,从而起到维持整个发酵微生态体系稳定性的作用[19-20]。基于这一点,进一步体现出RT高多样性带来的群落生态稳定性特点。从平均聚类系数上看,CRT稍高于AT,并且这两个子群的平均聚类系数明显高于其他子群,显著高于RT,表明CRT和AT中的细菌在各自子群内相比其他子群中的细菌关联程度更加紧密,具有更强的共存和相互协作性。此外,通过计算各子群在网络水平的拓扑系数,可以看出AT和CRT的平均路径长度和网络直径明显低于其他子群,而网络密度明显高于其他子群,进一步说明CRT和AT内的微生物相互作用和联系更加紧密。此外,从模块化程度看,AT最高,表明AT与其他子群相比,区系化更强。

图5 子群落间共存网络关系图Fig. 5 Network showing co-occurrence patterns among sub-communities
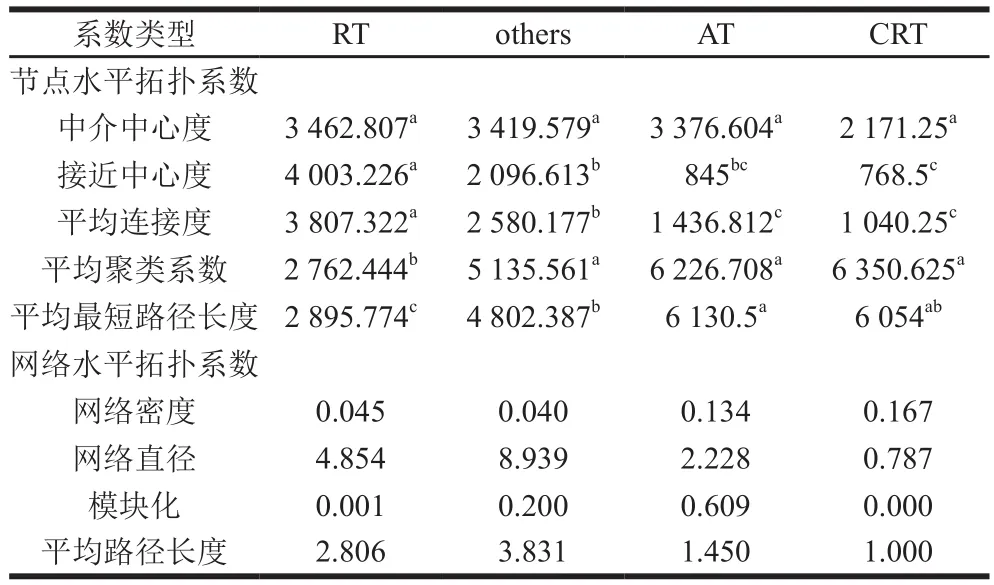
表2 各子群在共存网络中的拓扑系数Table 2 Topological coefficients of three sub-communities in co-occurrence network
2.4 群落演替与内源理化因子间关系
酒醅内源性理化因子,如乙醇、水分、乳酸、乙酸含量以及温度都会因发酵过程微生物代谢方式或活性的改变而发生改变,进而影响微生物群落的组成。为进一步理解各子群的演替与内源理化因子间关系,上述5个理化因子(表3)分别与各子群OTU相对丰度进行CCA(图6)。结果显示,酒醅内水分含量与AT演替显著相关(P<0.05,R2=0.09),AT由D1向D7的演替变化与水分含量呈正相关,随后从D7向D8的演替与水分含量呈负相关,表明AT与酒醅水分含量关系密切。乙酸与RT演替显著相关(P<0.05,R2=0.05),且整个发酵过程D1~D8,乙酸含量与RT呈负相关,表明RT的改变可能对乙酸含量影响较大。乙醇和乳酸含量与CRT演替显著相关(P<0.01,R2=0.52),且CRT从D1到D7的演替与温度、乙醇和乳酸含量呈正相关,从D7到D8的演替与温度呈正相关,与乙醇和乳酸含量呈负相关。此外,CRT的CCA模型R2至少是AT和RT的CCA模型R2的5 倍,表明CRT微生物对内源环境因子的敏感性远高于其他微生物,因此发酵环境的改变最易影响CRT微生物的组成结构,CRT的改变再对整个群落产生影响。对废水、有机物、饮用水、农林和土壤等微生态环境的微生物群落研究曾发现[19,20,35,47-48],CRT环境中如同微生物种子库,其种群密度会因环境条件的改变迅速扩大或消耗殆尽,可作为关键微生物调节整个群落结构,维持微生物间生态稳定性;同时,某些CRT微生物可作为环境改变的信号,指示具体影响微生物动态变化的物理、化学以及生物驱动力。在本研究基础上,可进一步探索CRT微生物,为调节、合理优化发酵体系中微生物结构另辟蹊径。
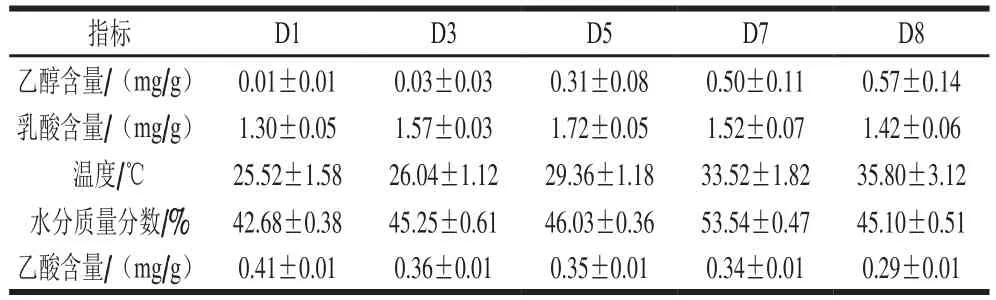
表3 堆积发酵不同时段酒醅内源理化指标Table 3 Physicochemical parameters of fermented grains at different stacking fermentation periods
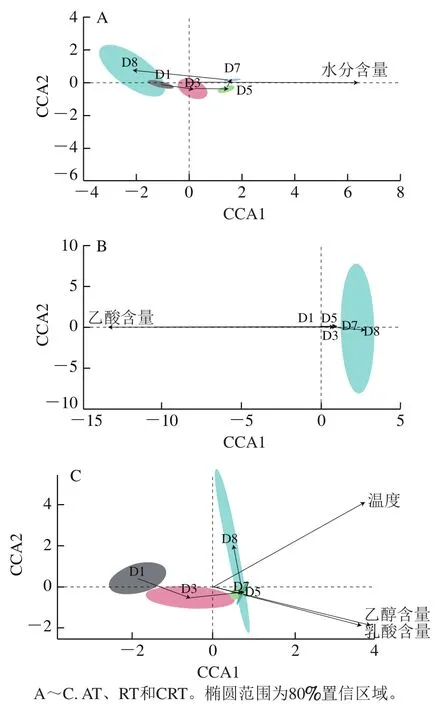
图6 各子群演替模式与内源理化因子间CCA排序图Fig. 6 CCA ordination describing the relationships between physicochemical factors and succession patterns of three sub-communities
3 结 论
本研究利用高通量测序技术,并结合一定的数理统计手段,系统阐释了AT、RT和CRT三类细菌群在酒醅堆积发酵过程中对WT演替的作用和影响,并得出相关结论:AT和CRT具有相似的演替模式,且AT决定WT的演替趋势和方向,CRT对WT的演替起到一定推动作用;WT演替速率主要由AT和CRT决定,WT物种积累数的缩减主要来自于RT的快速缩减;RT在WT中的演替相对独立,并且是维持发酵过程微生态体系稳定性的重要菌群,体系中环境的改变最易影响CRT组成结构,致使CRT对总群落结构产生影响。
