双水相萃取结合硼酸亲和吸附分离纯化辣根过氧化物酶
2022-02-16吴雅娇邢又元吴嘉聪李媛媛
吴雅娇,邢又元,韩 娟*,吴嘉聪,李媛媛,王 赟,*
(1.江苏大学化学化工学院,江苏 镇江 212003;2.江苏大学食品与生物工程学院,江苏 镇江 212003;3.江苏大学京江学院,江苏 镇江 212003)
辣根过氧化物酶(horseradish peroxidase,HRP,E.C.1.11.1.7)作为一种过氧化物酶,是由糖蛋白和褐铁卟啉组成的植物糖蛋白[1]。HRP是以过氧化氢为电子受体催化底物氧化的酶[2],目前已广泛用于化学工程[3]、环境保护[4-5]、突变育种[6]、医学诊断[7]和食品加工[8]等领域。辣根作为一种长年生香草,是HRP的主要来源。目前,HRP的纯化技术一直集中在各种色谱技术上,例如苯基琼脂糖凝胶CL-4B上的疏水相互作用色谱法[9]、伴刀豆球蛋白A琼脂糖亲和色谱法[10]、膨胀床离子交换色谱法[11]以及透析-凝胶过滤色谱法[12]等。然而,色谱技术既费时又昂贵,大规模应用受到限制。因此,开发一种操作简单、成本低和选择性好的纯化HRP的新方法备受关注。
双水相萃取已被证实是一种将分离、浓缩和纯化集成的单元操作。双水相体系(aqueous two-phase system,ATPS)是指将两种互溶的物质相混合,增加体系中物质的浓度达到(或超过)临界浓度后,原本互溶的均一体系会分成有明显界限的两相体系。双水相萃取具有成本低、生物相容性好和易于扩大等优点。目前,聚合物-盐ATPS已广泛用于分离纯化蛋白质[13],例如木瓜蛋白酶[14]、菠萝蛋白酶[15]、卵清蛋白[16]和纤溶蛋白酶[17]。然而,尽管双水相体系在分离纯化蛋白质方面有一定优势,但目标蛋白质选择性差和聚合物价格昂贵仍然是其工业应用的主要限制。
近年来,磁性氧化石墨烯(graphene oxide@Fe3O4,GO@Fe3O4)在分离纯化蛋白质领域表现优异,引起了广大学者的关注。其中,GO比表面积大[18],易于修饰和生物相容性好[19],同时Fe3O4具有重复利用性,可降低分离成本[20]。因此,GO@Fe3O4纳米材料是分离和纯化蛋白质的理想载体[21]。树枝状聚合物聚乙烯亚胺(polyethyleneimine,PEI)具有丰富的官能团,可作为GO@Fe3O4的主要支架增强材料和蛋白质的结合强度。基于此,本实验利用一种“多价”苯硼酸亲和磁性石墨烯复合材料(d-PBA-GO@Fe3O4@PEI)作为进一步纯化糖蛋白的分离材料。一方面,磁性材料表面连接了苯硼酸基团,苯硼酸与糖类化合物上的邻二羟基或间二羟基能形成可逆硼酸酯键,这种酯键会受外界环境pH值的影响,弱碱性条件下形成,酸性条件下解离,使得磁性材料与糖蛋白具有特异性吸附;另一方面,树状聚合物PEI可以放大硼酸配体的数量,从而增加对糖蛋白的结合亲和力。
基于此,本实验将双水相萃取与亲和吸附相结合,建立一种集成化方法用于植物中HRP的分离纯化。首先以9种模型化合物为例,优化两步双水相萃取体系的最佳条件,接着加入“多价”苯硼酸亲和磁性石墨烯复合材料进一步纯化HRP,构建适用于HRP分离纯化的集成化方法。最终将该集成化方法用于植物辣根粗提液中,实现HRP的分离纯化。该集成化方法具有优异的分离纯化性能,与传统色谱技术相比,具有操作简单和成本低的优势,对植物糖蛋白的工业化生产具有良好的前景。
1 材料与方法
1.1 材料与试剂
17R4聚环氧丙烷-聚环氧乙烷-聚环氧丙烷(polypropylene oxide-polyethylene oxide-polypropylene oxide,PPO-PEO-PPO,相对分子质量2 700)、(NH4)2SO4(纯度≥99.0%)、磷酸氢二钾(纯度≥99.0%)、柠檬酸钾(纯度≥99.5%)、浓硫酸(纯度98.0%)、高锰酸钾和和叶绿素a(chlorophyll-a,CHL,纯度≥85%) 国药集团化学试剂有限公司;HRP(酶活力>150 U/mg) 麦克林生化科技有限公司;细胞色素C(cytochrome C,CYT,来自牛心,纯度≥95.0%)、牛血清白蛋白(bovine serum albumin,BSA,纯度96.0%)、花青素(anthocyanin,ANT,纯度95.0%)、胡萝卜素(carotene,CAR,纯度≥95.0%)、黄芪多糖(astragalus polysaccharides,ASP,纯度>98.0%)、蔗糖(sucrose,SUC,纯度≥99.0%)、果糖(fructose,FRU,纯度99.0%)、石墨粉、六水合氯化铁、氨水、七水合亚硫酸铁、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺、氰基硼氢化钠、4-甲酰基苯基硼酸 阿拉丁生化科技股份有限公司。所有化学试剂无需进一步纯化即可使用。
1.2 仪器与设备
DF-101S恒温磁力搅拌器 河南省巩义予华仪器有限公司;UV-2450紫外-可见分光光度计 日本岛津公司;RX-DD7折光率仪 日本爱拓公司;DDS 307A电导率仪上海仪电科学仪器股份有限公司;PHS-3C pH计 上海精密科学仪器有限公司。
1.3 方法
1.3.1 d-PBA-GO@Fe3O4@PEI的制备
根据本课题组前期的工作合成d-PBA-GO@Fe3O4@PEI[22],得到的材料用磁铁吸出,用水和乙醇洗涤,40 ℃干燥备用。
1.3.2 浊点的测定
制备一系列不同浓度的17R4纯溶液及加入盐(K3C6H5O7/(NH4)2SO4/K2HPO4,质量分数1%)的混合溶液。将数字温度计放入盛装上述溶液的试管中并在恒温水浴锅中加热,直到溶液变浑浊为止。然后将样品从水浴锅中取出并在室温下冷却。通过观察捕捉浊度消失的突变点,以获得溶液的浊点。温度的误差控制在0.1 ℃以内,将3 次测定结果取平均值即为浊点。
1.3.3 液液相平衡的测定
通过滴定法测定17R4-盐ATPS的液液相平衡数据。首先,将质量分数已知的聚合物溶液加入到锥形烧瓶中,并浸入25 ℃恒温水浴中。然后,将质量分数已知的盐溶液滴入锥形瓶中,直到澄清溶液变浑浊为止。将去离子水滴加到烧瓶中以得到澄清溶液,并重复该过程。通过称量法计算混浊点17R4(w1)和盐(w2)的质量分数。
1.3.4 9种目标物在集成化方法中的分配
首先,将一定量的9种模型化合物(CHL、HRP、CYT、BSA、ANT、CAR、ASP、SUC、FRU)加入到17R4-盐ATPS中,相平衡后,3种多糖分布到富盐相中,而其他模型化合物富集在17R4富集相中,从而实现了糖类物质的去除。其次,将柠檬酸钠电解质溶液添加至富17R4相中,调节其相间电位差后控温形成17R4-柠檬酸钠ATPS,此时蛋白质分离进入盐富集相中,色素仍然保留在17R4富集相中,从而达到去除色素的目的。然后,在上述富盐相中加入一定量的d-PBA-GO@Fe3O4@PEI吸附剂,调节最佳吸附条件,实现HRP与d-PBA-GO@Fe3O4@PEI的亲和吸附,而其他两种杂质蛋白仍然保留在水相中。最后,调控吸附条件实现洗脱HRP。
1.3.5 植物辣根中HRP的分离纯化
首先洗净辣根(25 g),然后在4 ℃加入100 mL预冷的50 mmol/L磷酸盐缓冲液(phosphate buffered saline,PBS,pH 7.5),12 000 r/min匀浆5 次。抽滤后,将上述PBS添加至滤液中至200 mL。在冰水浴条件下,用超声波(40 kHz)将溶液超声处理3 次,每次20 min。摇动后,将提取物4 ℃、8 000 r/min离心15 min,得到一定量的辣根粗提取物。将一定量辣根粗提物加入到两步ATPS进行初级纯化,接着用d-PBA-GO@Fe3O4@PEI进一步亲和吸附HRP,最后得到纯化后的HRP。
目标物的酶活力回收率(Ye)、蛋白分布系数(Kp)、纯化因子(PF)、分配系数(K)和提取效率(E)的计算如式(1)~(5)所示:

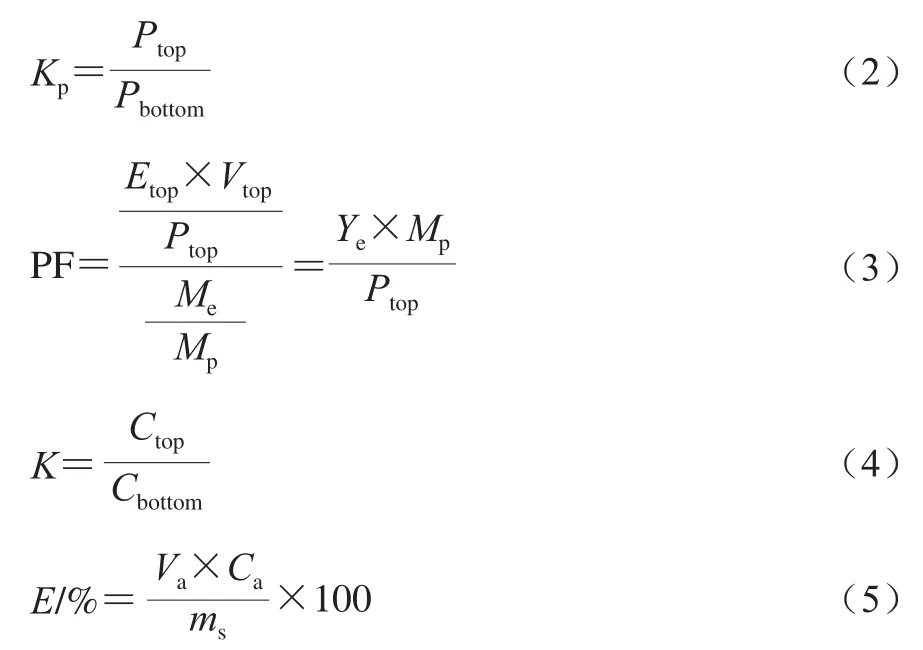
式中:Etop、Vtop和Me分别为上相的酶活力/(U/mL)、上相的体积/mL和初始总酶活力/(U/mL);Mp、Ptop和Pbottom分别为初始蛋白质量浓度/(g/mL)、上相和下相中的蛋白质量浓度/(g/mL);Ctop和Cbottom分别为目标物在上相和下相中的平衡质量浓度/(g/mL);Ca、Va和ms分别为富集相中的目标物质量浓度/(g/mL)、富集相的体积/mL和目标物的初始添加量/g。
1.3.6 目标物的测定
用Micro BCATM蛋白测定试剂盒测定HRP、BSA和CYT的含量[23];ASP、PUP和FRU含量通过苯酚-硫酸法测定[24]。
CAR测定:通过将25 mg CAR溶于少量三氯甲烷并用石油醚稀释至100 mL,可得到250 μg/mL的CAR母液。然后通过稀释CAR母液获得不同质量浓度(5、10、20、30、40、50、60 μg/mL)的CAR标准溶液,并通过测量450 nm波长处的吸光度绘制CAR的标准曲线。用石油醚稀释后,在450 nm波长处测定样品溶液的吸光度。
ANT测定:将ANT(20 mg)溶于10 mL甲醇中,获得ANT母液,并通过稀释母液获得不同质量浓度(0.031 25、0.062 5、0.25、0.5、1 mg/mL)的标准溶液。然后将2.5 mL硫酸添加至0.5 mL标准溶液中,并通过分光光度计在500 nm波长下测定溶液吸光度以绘制标准曲线。样品溶液用甲醇稀释,并根据上述程序测定吸光度。
CHL测定:将CHL(10 mg)溶于100 mL乙醇中,以制备不同质量浓度(10、20、30、40、50、60 μg/mL)的标准溶液。在652 nm波长处测定吸光度以绘制标准曲线。用乙醇稀释后测定样品溶液的吸光度。
1.3.7 酶活力测定
将一定质量浓度HRP溶液0.1 mL转移至底物溶液中,该底物溶液包含1.4 mL 4-氨基安替吡啉溶液和1.5 mL H2O2溶液。随后,每1 min记录一次510 nm波长处溶液的吸光度,直到每分钟内吸光度的增加达到稳定值。HRP活力通过式(6)计算:
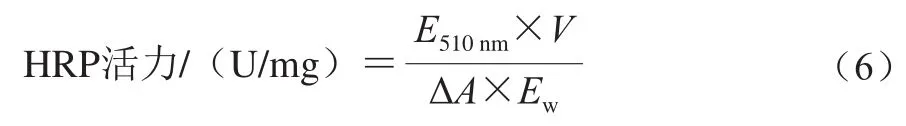
式中:E510nm为510 nm波长处每分钟吸光度的增加量;V为反应液的总体积/mL;ΔA为6.58,表示1个单位HRP能在1 min内使吸光度提高6.58;Ew为0.1 mL酶溶液中酶质量/mg。
1.4 数据统计与分析
在进行3 次重复实验后,使用Excel 2010和Origin 9.0软件进行统计分析,通过t检验进行单因素方差分析,后用Duncan法进行差异显著性比较,P<0.05,差异显著。作图使用Origin 9.0软件。
2 结果与分析
2.1 17R4浊点的分析
温敏聚合物浊点研究是构建温敏聚合物-盐ATPS的依据。17R4含量与浊点的关系如图1所示,浊点会随着17R4质量分数的增加而降低。由于胶束-胶束相互作用增加,胶束浓度增加从而使自由水成分相对减少,胶束则更容易团聚。浊点在0.1% 17R4时达到最高(81 ℃),在10% 17R4时达到最低(25.5 ℃)。为考察盐对浊点的影响,向17R4溶液中加入一定量的3种盐((NH4)2SO4、K3C6H5O7、K2HPO4),并分别测量其浊点变化。结果显示3种盐均会降低17R4的浊点。受盐析能力的影响[25],体系中的盐会与聚合物竞争水分子,使胶束脱水并增强胶束间的作用力。因此,盐可以降低聚合物溶液的浊点温度。此外,每种盐对浊点的影响也不同,这3种盐改变浊点的能力排序如下:(NH4)2SO4>K3C6H5O7>K2HPO4。
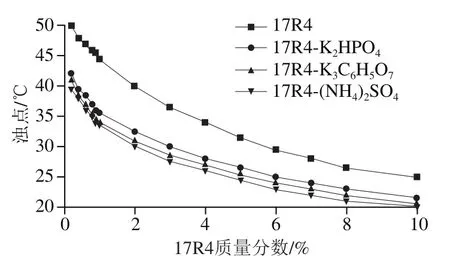
图1 17R4溶液的浊点与含量及盐的关系Fig. 1 Relationship between cloud point and concentration of 17R4 solution with different salts
2.2 17R4-盐ATPS的液液相平衡行为
实验测定了17R4-盐ATPS的液液相平衡组成,如表1所示。当ATPS分相后,17R4富集在ATPS的上相,盐富集在ATPS的下相。增加盐或聚合物的用量均使系线长度延长,有利于ATPS的液液相分离。通过实验还发现,当聚合物质量分数在7%~8%,盐质量分数在13.2%~13.7%时的分相效果较好。从图2可以看出,聚合物17R4质量分数8%的斜率均大于7%,相稀释度更小,分相效果更佳。图1表明盐对17R4相分离的影响顺序为(NH4)2SO4>K2HPO4>K3C6H5O7。由于盐的水合作用比较强,与聚合物争夺水分子,所以发生了相分离。对于此ATPS,无机盐比有机盐的分相效果强,(NH4)2SO4的盐析能力最好。因此,实验选择8% 17R4-13.2%~13.7% (NH4)2SO4ATPS作为初级分离体系。
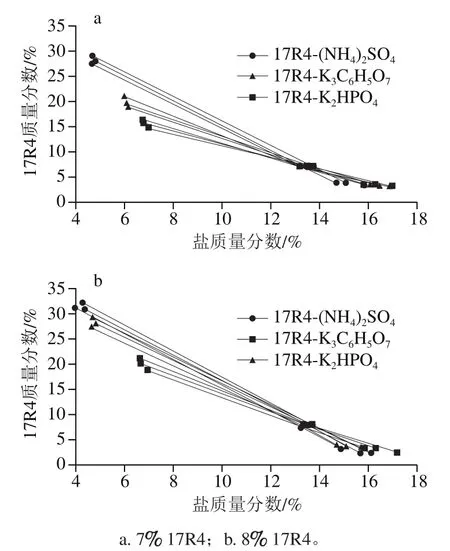
图2 聚合物种类在25 ℃条件下对双水相体系液液相平衡组成的影响Fig. 2 Effect of polymer type on the liquid-liquid equilibrium composition of two-phase aqueous system at 25 ℃
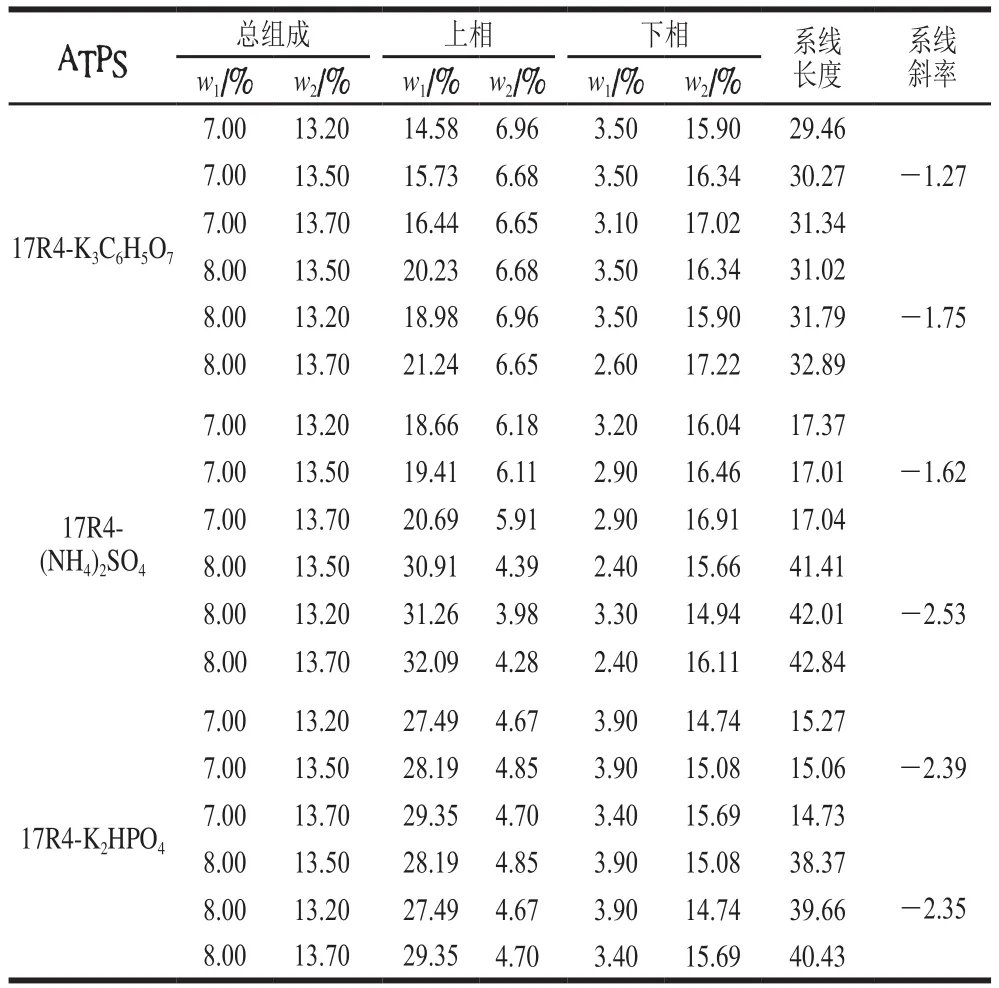
表1 17R4(w=7%/8%)-K3C6H5O7/(NH4)2SO4/K2HPO4ATPS在25 ℃的液液相平衡组成Table 1 Lliquid-liquid equilibrium data for 17R4 (w =7%/8%)-K3C6H5O7/(NH4)2SO4/K2HPO4 ATPSs at 293.15 K
2.3 9种模型化合物在17R4-盐ATPS中的分配
由于植物中存在各种化学成分,有效分离和纯化植物中的特定成分尤为困难。为选择用于选择性分离HRP的最佳ATPS,实验选择3种模型化合物(包括多糖、色素和蛋白质)研究ATPS作为初级纯化步骤的效果。实验探究HRP、BSA、CYT、ASP、SUC、FRU、CHL、ANT和CAR在17R4-盐ATPS中的分配情况。实验发现糖类物质主要分布在盐富集相,而色素和蛋白质则倾向于分布在温敏聚合物相。当固定17R4质量分数为8%时,3种盐对9种模型化合物的分离效率影响顺序为(NH4)2SO4>K2HPO4>K3C6H5O7。因此,选择8% 17R4-(NH4)2SO4ATPS作为分离最佳体系。(NH4)2SO4质量分数的改变对9种模型化合物分离效果见图3。(NH4)2SO4质量分数增加会导致其盐析作用增强,从而将目标物驱动分配到聚合物富集相。但(NH4)2SO4质量分数过高也会导致盐富集相的黏度降低,从而导致传质效率和提取效率的降低。因此,实验选择8% 17R4-13.2% (NH4)2SO4ATPS作为HRP的初步纯化体系。此时9种模型化合物在聚合物富集相的最佳提取效率分别为:(88.88±2.82)%(HRP)、(75.78±2.76)%(BSA)、(74.16±2.64)%(CYT)、(94.96±2.32)%(CHL)、(94.96±3.12)%(ANT)、(87.05±3.46)%(CAR)、(7.64±3.14)%(SUC)、(10.12±2.79)%(ASP)和(2.01±1.52)%(FRU)。
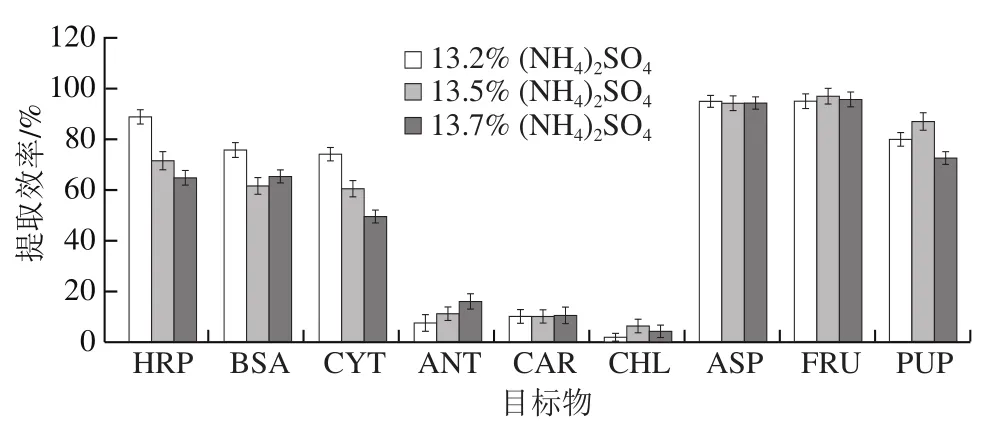
图3 9种目标物在8%17R4-(NH4)2SO4ATPS中的分配效果Fig. 3 Partition efficiency of nine target compounds in 8%17R4-(NH4)2SO4 ATPS
2.4 17R4-Na3C6H5O7 ATPS进一步分离蛋白质和色素
将9种模型化合物分配到17R4-(NH4)2SO4ATPS后,蛋白质和色素分离富集到温敏聚合物17R4相中。为了进一步分离色素和蛋白质,将电解质添加到ATPS中,然后将蛋白质反萃取到富盐相中。根据先前关于电解质调节对ATPS中蛋白质分布影响的研究,通过添加电解质可以产生不同的静电势(相间势)[26]。由于蛋白质的电荷状态取决于pH值,可以通过操纵电解质和pH值调节蛋白质的分配。因此,研究Na3C6H5O7、Na2SO4和NaCl 3种电解质对蛋白质分配的影响。考虑到盐析作用的影响,控制体系中电解质的浓度为100 mmol/L以消除盐析作用的影响,同时具有最大化相间电位的作用[27]。为充分了解外加电解质对体系蛋白质分配的过程影响,以加入Na3C6H5O7为例进行详细介绍。对疏水性的凝聚相具有较强的亲和性,故向体系中加入Na3C6H5O7时,主要分配于体系下相,从而在两相间产生相间电位差。在此电势的作用下,体系中带正电的物质会富集于体系的凝聚相,同时,体系中带负电物质则被排斥到体系稀释相。蛋白质的等电点大多为酸/中性左右,而当体系pH值为8.0时,蛋白质带负电荷。此时,加入Na3C6H5O7所产生的相间电位会对蛋白质产生排斥作用,进入稀释相。另外,当体系pH值为7(接近等电点)时,蛋白质几乎不带电,加入Na3C6H5O7产生的相间电位并不会对蛋白质在两相间分配以及蛋白质回收率产生显著影响。如图4所示,在实际实验中,加入Na3C6H5O7对蛋白质去除率造成的影响均与上述结论相符,从而验证了前文论述的正确性,并证实了借助调节体系pH值与外加电解质调节荷电物质(如蛋白质)在两相间分配的可行性。另外,还向体系中加入Na2SO4和NaCl以研究和Cl-对体系蛋白质和色素回收率的影响,结果如图4所示。可知不同电解质离子的疏水性序列如下:该序列与文献所述相符,从而论证了上述理论的准确性[28]。因此,当Na3C6H5O7添加到ATPS中时,大多数蛋白质从富含17R4的相进入富含盐的相,因此通过这种反萃取操作实现了蛋白质和色素的分离。富盐相中HRP、BSA和CYT 3种蛋白质的提取效率分别为(75.43±1.26)%、(73.80±1.73)%和(70.38±2.33)%,而盐富集相中ANT、CAR和CHL3种色素的提取效率分别为(1.07±0.33)%、(1.37±0.21) %和(2.11±0.38)%。此步骤成功实现了从蛋白质中去除色素的目的。
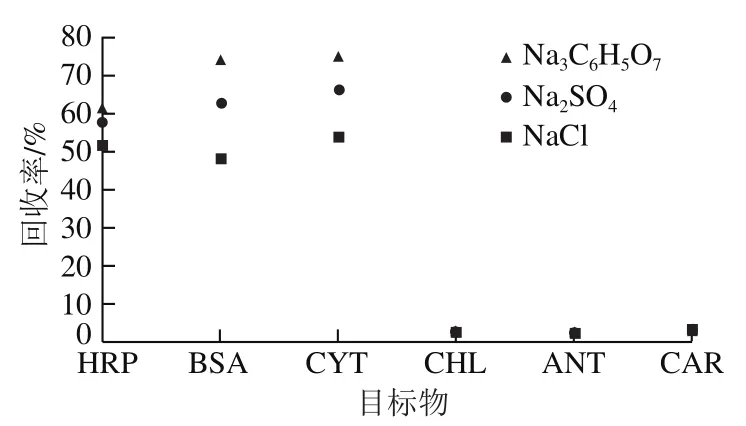
图4 外加电解质对体系蛋白质和色素回收率的影响Fig. 4 Influence of added electrolyte on recoveries of protein and pigment
2.5 苯硼酸亲和吸附材料进一步纯化HRP
课题组前期合成了一种树枝状“多价”苯硼酸亲和磁性石墨烯复合材料d-PBA-GO@Fe3O4@PEI用于HRP的分离,选择性分离效果令人满意[22]。GO比表面积大,且表面含有丰富的官能团,拥有较强的生物相容性,可以被其他功能聚合物修饰改性。GO的这些优异特性为蛋白酶的吸附提供了空间以及活性位点,同时,Fe3O4具有特定的磁性结构和可回收特性,可重复利用,从而降低蛋白质分离的成本[29]。树状聚合物PEI用于增强磁性材料与糖蛋白的结合强度。由于其独特的特性,丰富的官能团和易于修饰,树枝状PEI被选作磁性材料的主要支架。而且,PEI链上的丰富氨基还可以改善材料的亲水性,并有助于减少非糖蛋白的吸附。同时,还在磁性材料表面连接了苯硼酸基团,使材料与糖蛋白特异性吸附。PEI已被证明是一种出色的支架,可以放大硼酸配体的数量,从而增加对糖蛋白的结合亲和力。根据前期的讨论结果[22],d-PBA-GO@Fe3O4@PEI吸附HRP的最佳条件为:HRP质量浓度0.9 mg/mL、吸附pH 8.0、吸附温度35 ℃以及吸附时间60 min。当吸附完全后,磁性回收吸附剂和酶,加入一定量的100 mmol/L乙酸溶液,悬浮1 h,用以洗脱吸附剂上的HRP。通过以上两步ATPS纯化和d-PBA-GO@Fe3O4@PEI进一步分离纯化HRP后,计算得到HRP的提取效率为(70.30±2.08)%,BSA和CYT的提取效率分别为(8.11±0.35)%和(7.07±0.75)%,成功实现HRP的选择性分离。
2.6 辣根中HRP的分离纯化
通过将构建的集成化方法用于从辣根粗提液中选择性分离HRP,验证该集成方法对HRP分离纯化的有效性。结果表明,纯化因子为1.92,比活力为28.93 U/mg,萃取回收率为(65.20±1.15)%。此外,通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfatepolyacrylamide gel electrophoresis,SDS-PAGE)证明纯化效果。如图5所示,辣根粗提液中存在许多蛋白质杂质。17R4-(NH4)2SO4ATPS提取后,蛋白质杂质减少,如SDSPAGE的第L道所示。当加入d-PBA-GO@Fe3O4@PEI进一步吸附分离后,洗脱液中有明显的HRP条带,证明该集成化方法在从辣根中分离和纯化HRP方面显示出良好的性能。
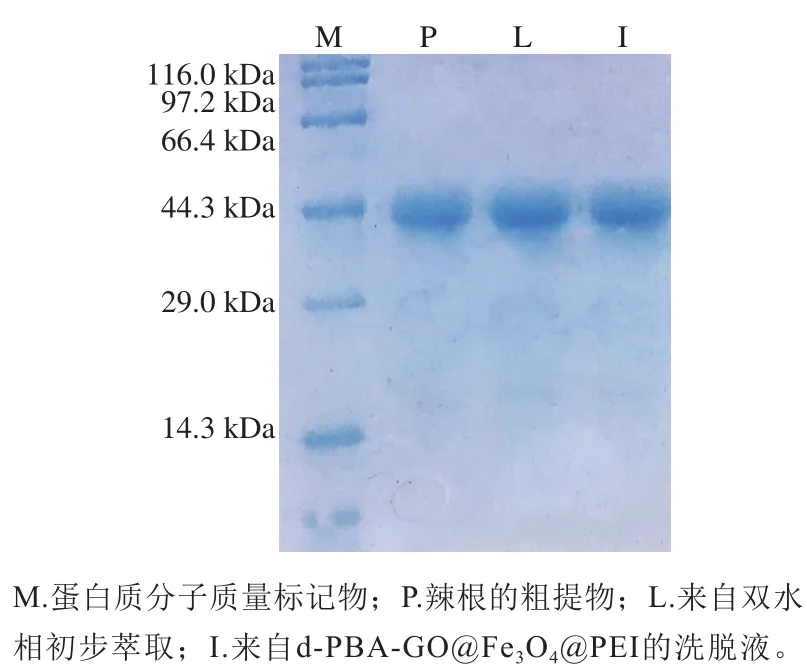
图5 辣根粗提取物中提取HRP的SDS-PAGE分析Fig. 5 SDS-PAGE analysis of horseradish peroxidase extracted from crude horseradish extract
2.7 纯化后HRP的结构分析
用红外光谱和圆二色谱分析纯化后HRP的结构变化。HRP的二级蛋白质结构出现在1 649 cm-1和1 541 cm-1的酰胺带上[30]。从图6a可以看出,纯化后HRP和商品化HRP的酰胺带峰位置相同,纯化后HRP和商品化HRP的其他峰位也基本相似。如图6b所示,作为HRP的共同特征,208 nm和222 nm处的负峰是典型的α-螺旋结构,而大约196 nm处的正峰属于β-折叠结构。通过对比二者的光谱结构,可以充分证明本实验的集成化方法对HRP结构没有影响。
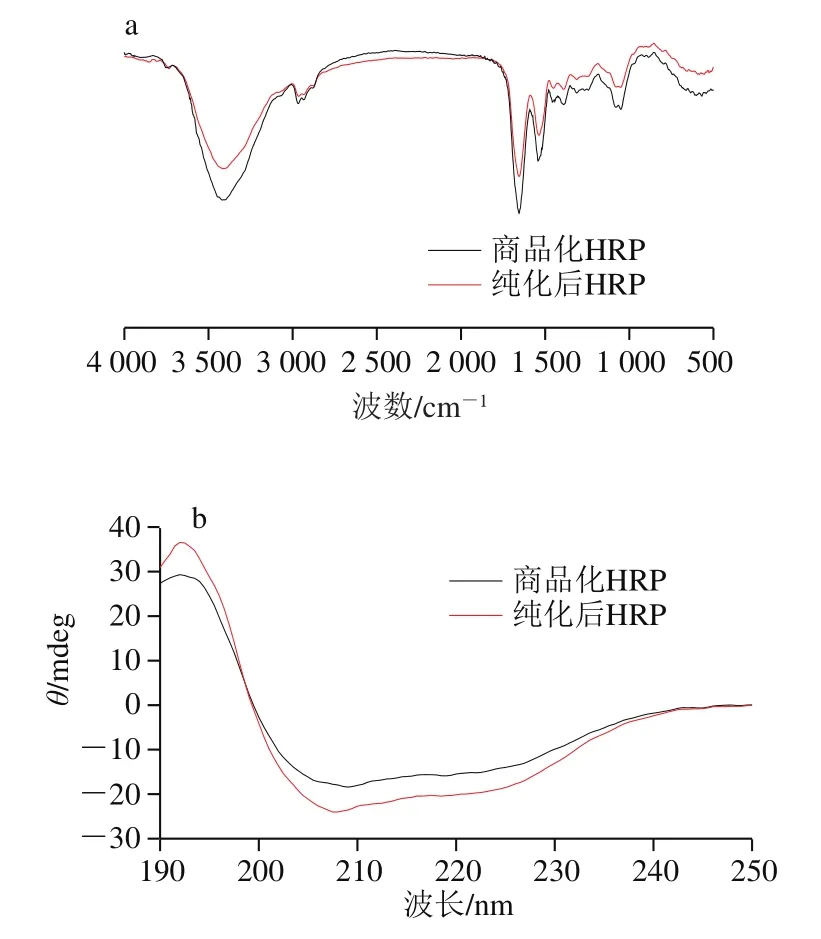
图6 纯化后及商品化HRP的红外光谱(a)和圆二色谱(b)图Fig. 6 FT-IR spectra (a) and CD spectra (b) of purified HRP and commercial HRP
3 结 论
本实验构建了17R4-(NH4)2SO4/K2HPO4/K3C6H5O7温敏双水相萃取体系,探讨并优化了用于纯化蛋白质的条件;模拟辣根中未知的物质和环境研究9种目标物在双水相体系种的分配情况,发现采用17R4-(NH4)2SO4ATPS在辣根粗酶液中初步分离纯化HRP可得到(75.43±1.26)%的提取效率。最后在粗提液中加入d-PBA-GO@Fe3O4@PEI磁性材料选择性吸附HRP,纯化后的HRP纯化因子为1.92,比活力为28.93 U/mg,萃取回收率为(65.20±1.15)%,得到了较为满意的纯化结果。
