利托那韦过饱和溶液中相分离研究*
2021-03-08马奇三赵燕娜赵玉萍洪建华王正平韩军
马奇三,赵燕娜,赵玉萍,洪建华,王正平,韩军
(1.聊城大学生物制药研究院,聊城 252000;2.南昌航空大学环境与材料学院,南昌 330063)
目前,随着组合化学和高通量筛选技术的发展,难溶性候选化合物的数量急剧增加。据统计,上市药品中约40%水溶性较差,而在研阶段候选药物难溶性比例高达90%[1]。因此,改善难溶性药物溶解度是制剂开发中最具挑战的问题之一[1],一些提升药物溶解度的制剂技术也因此应运而生,固体分散即为其中一种较有效的方式。固体分散体中的药物往往以无定型或分子状态分散在载体材料中,熔融法和溶剂挥发法是最常用的两种方法[2-3]。由于药物无定型状态相比于晶态是高能态,溶解不受晶格能束缚,所以有较高溶解度,进而可提高药物生物利用度[4-5],并由此产生处于亚稳态的过饱和溶液[6-8]。合理运用辅料可以在一定程度上阻止药物成核和晶体增长,确保药物在存储期间保持无定型和阻止无定型药物溶出后重结晶[9-10]。但迄今为止利用固体分散技术制备的药物却仍屈指可数,根本原因是固体分散体本质上不稳定,高能态无定型药物更倾向于转化为低能态难溶结晶形式,这使得固体分散体优势大打折扣。由难溶性药物和水溶性高分子聚合物组成的非晶固体分散体(amorphous solid dispersion,ASD)作为过饱和药物传递系统,已被证明在溶于水的过程中能够产生第二分散相[11-12],对于第二分散相形成过程中的相行为目前知之甚少。
人体免疫缺陷病毒(human immunodeficiency virus,HIV)蛋白酶抑制剂利托那韦,是艾滋病“鸡尾酒”疗法的重要组分,在控制病毒传播和控制病毒变异产生抗药性方面疗效显著[13],临床可单独使用,或与其他抗逆转录药物联用[14]。利托那韦水溶性较差,已被证实在水介质中可形成第二分散相[15]。笔者在本研究选用利托那韦为模型药物,通过溶剂转移法考察过饱和状态下利托那韦胶体分散体粒径变化和结晶趋势,利用动态光散射、消光(浊度)、芘荧光探针、偏光显微镜全面表征其过饱和溶液相分离及结晶过程。研究结果可为筛选具有优异结晶抑制特性的聚合物提供新思路,并有助于指导新型药用辅料开发。
1 仪器与试药
1.1仪器 Nano ZSP激光粒度仪(英国马尔文仪器公司),F-7000荧光分光光度计(日本日立公司),U-3900H紫外分光光度计(日本日立公司),Axio lab A1偏光显微镜(卡尔蔡司股份公司)。
1.2试药 利托那韦原料药(印度HETERO DRUGS LIMITED,含量:99%,批号:RI17100047),芘(麦克林试剂,含量:99%,批号:C10561824),磷酸二氢钾(分析纯,天津市致远化学试剂有限公司,含量≥99.5%,批号:20108012079),氢氧化钠(分析纯,烟台远东精细化工有限公司,含量≥96%,批号:20181101),甲醇(色谱纯,阿达玛斯试剂,含量≥99.9%,批号:P1346953),水为自制超纯水。
2 方法与结果
2.1过饱和溶液的制备与第二分散相观察 通过溶剂转移法制备利托那韦过饱和溶液。取利托那韦原料药50 mg,溶于甲醇5 mL,作为储备液备用。移取储备液0.5 mL,滴入pH值6.8磷酸盐缓冲液50 mL中(37 ℃,300 r·min-1),在药物滴加的过程中,磷酸盐缓冲液由澄清变浑浊,呈现胶体溶液特有的蓝色乳光,最终形成理论浓度100 μg·mL-1利托那韦过饱和溶液。取该过饱和溶液20 μL,滴于载玻片上,偏光显微镜观察,可见亚微米级非双折射的类液体状球形小液滴,见图1。由前人研究可知,这些小液滴是当溶液中药物溶度超过其无定型溶解度时经液-液相分离形成的第二分散相,该相是一种富含药物的类液体状的液滴[15-16]。
2.2第二分散相形成的临界浓度测定 为测定第二分散相形成的临界浓度,分别利用紫外分光光度计和动态光散射对该过程进行表征。通过溶剂转移法,向pH值6.8磷酸盐缓冲液50 mL中依次加入利托那韦甲醇储备液,获得浓度分别为5,10,15,20,25,30,35,40,45 μg·mL-1过饱和溶液,取相应浓度过饱和溶液适量,测其紫外消光和动态光散射粒径。其中,紫外吸收波长选择无药物紫外吸收干扰波段(如280 nm),以消光(extinction)作为评价指标。动态光散射则通过马尔文粒度仪(Nano ZSP,以173°散射角),以光强(count rate)作为评价指标。以消光或光强对浓度作图,结果见图2。由图2可知,紫外消光和光强结果一致,均在利托那韦浓度达到约37 μg·mL-1时产生突变,表明液-液相分离浓度,即第二分散相形成的浓度约37 μg·mL-1。

图1 利托那韦过饱和溶液偏光图
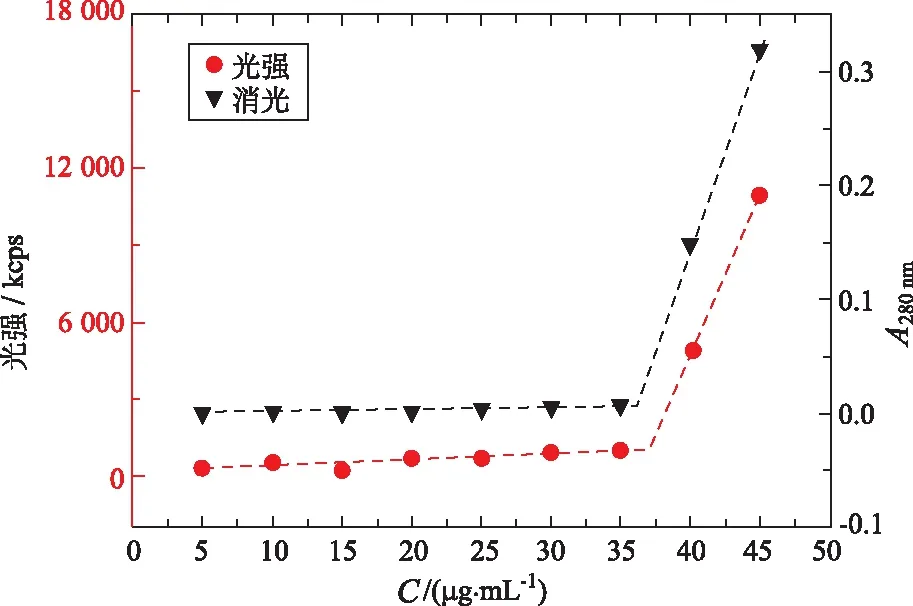
图2 利托那韦过饱和溶液相分离浓度曲线
2.3过饱和溶液中产生的第二分散相的性质考察 结合“2.2”项得到的相分离浓度,进一步考察相分离后形成的第二分散相性质。50 mL pH值6.8磷酸盐缓冲液37 ℃条件下300 r·min-1搅拌,每隔5 min采集一次光强和紫外消光数据,作为空白对照。20 min后,加入利托那韦甲醇储备液0.2 mL,获得浓度为40 μg·mL-1利托那韦溶液,继续每隔5 min采集一次光强和紫外消光数据。20 min后加入pH值6.8磷酸盐缓冲液50 mL,使其浓度稀释为20 μg·mL-1,继续每隔5 min采集一次光强和紫外消光数据,结果见图3。由图3可知,利托那韦过饱和溶液达40 μg·mL-1,会发生液-液相分离形成第二分散相,与“2.2”项结果一致。而当利托那韦过饱和溶液被稀释至20 μg·mL-1时,其消光和光强又骤降到接近0 μg·mL-1水平,说明液-液相分离形成的第二分散相可逆,即该富药液滴并非晶体,否则其无法在高于利托那韦平衡溶解度的溶液中溶解,消光和光强也不会骤降到如此低。但需要注意的是,利托那韦过饱和溶液再稀释后的消光和光强并没有回到与0 μg·mL-1一致的零点,而是略高于零点,推测溶液中存在少量未溶解的晶体[17]。另外在40 μg·mL-1浓度下,随着时间延长,利托那韦过饱和溶液消光和光强一直下降,推测可能是该溶液中富药液滴不稳定,药物结晶析出所致。
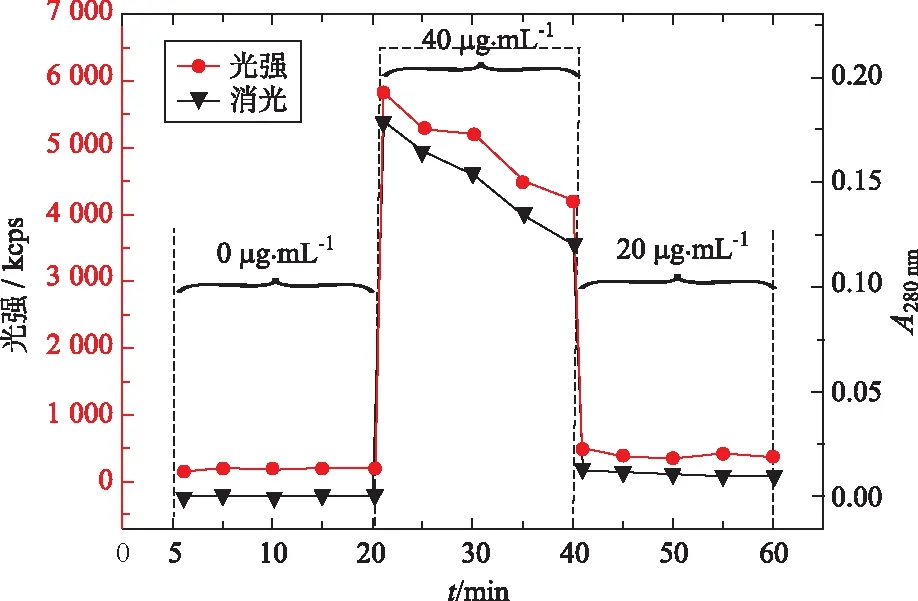
图3 浓度变化过程中利托那韦过饱和溶液光强和消光变化曲线
2.4第二分散相结晶情况考察 通过芘荧光探针进一步表征利托那韦过饱和溶液中第二分散相结晶过程,芘的荧光发射峰强度之比(I1/I3)对芘分子所在微环境的极性特别敏感,在药物结晶前,芘进入富药液滴极性较小的疏水环境中,而富药液滴结晶时,芘被排出晶格,进入极性大的水溶液,因此可以利用芘荧光探针法测定第二分散相富药液滴环境的极性变化和结晶过程[16]。称取芘1 mg,用二甲亚砜1 mL溶解,取25 μL加入pH值6.8磷酸盐缓冲液50 mL,使芘终浓度为0.5 μg·mL-1。取利托那韦储备液0.2 mL加入上述含芘磷酸盐缓冲液,利托那韦终浓度为40 μg·mL-1,37 ℃条件下300 r·min-1持续搅拌,每几分钟测一次荧光。荧光分光光度计激发波长为334 nm,375 nm处的发射峰为I1,386 nm处的发射峰为I3,以I1/I3对时间作图,结果见图4。由图4可知,加入利托那韦使I1/I3迅速减小,说明短时间内利托那韦过饱和溶液发生了相分离,并且产生了具有疏水核心的富药液滴,即第二分散相。随着时间延长,I1/I3又逐渐增大,说明部分液滴或液滴局部开始结晶,药物分子开始有序排列,芘被排出晶格,进入极性强的水环境中。
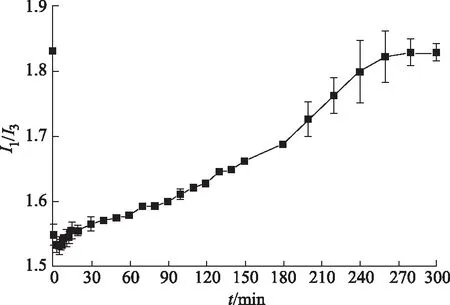
图4 I1/I3随时间变化曲线
2.5第二分散相粒径的变化考察 进一步考察第二分散相结晶过程伴随粒径变化,取利托那韦储备液0.2 mL,加入pH值6.8磷酸盐缓冲液50 mL中,利托那韦终浓度40 μg·mL-1,37 ℃,300 r·min-1持续搅拌,一定时间间隔取样,马尔文nano-ZSP粒度仪测粒径,采用后向散射探测器,探测角度为173°的散射光。从图5粒径随时间的变化曲线可以看出,第二分散相富药液滴初始粒径在约450 nm,随着时间延长,其粒径逐渐增大,到120 min,粒径增大到约1600 nm,结合原始数据,在120 min之前,第二分散相粒径的标准偏差较小,多分散指数(polydispersity index,PDI)<0.7,说明液滴粒径分布较集中。在120 min后,粒径仍呈增长趋势,但粒径标准偏差变得很大,同时PDI>0.9,甚至等于1,说明液滴粒径分布较宽,大颗粒和小颗粒同时存在。
2.6考察第二分散相在偏光显微镜下的变化 为直观观察过饱和溶液中第二分散相结晶情况,取利托那韦储备液0.2 mL,加入pH值6.8磷酸盐缓冲液50 mL中,利托那韦终浓度40 μg·mL-1,37 ℃条件下300 r·min-1持续搅拌,10,30,60,90,180,240 min取样,偏光显微镜观察,结果见图6。由图6可见,从10~60 min,过饱和溶液第二分散相的形成,第二分散相粒径逐渐变大,并且液滴在偏光下逐步显现出晶体的光学特征,晶核逐渐形成,90 min可以看到晶体的初步形成,180~240 min,可以观察到晶体生长,小晶体进一步生长变为更大晶体。
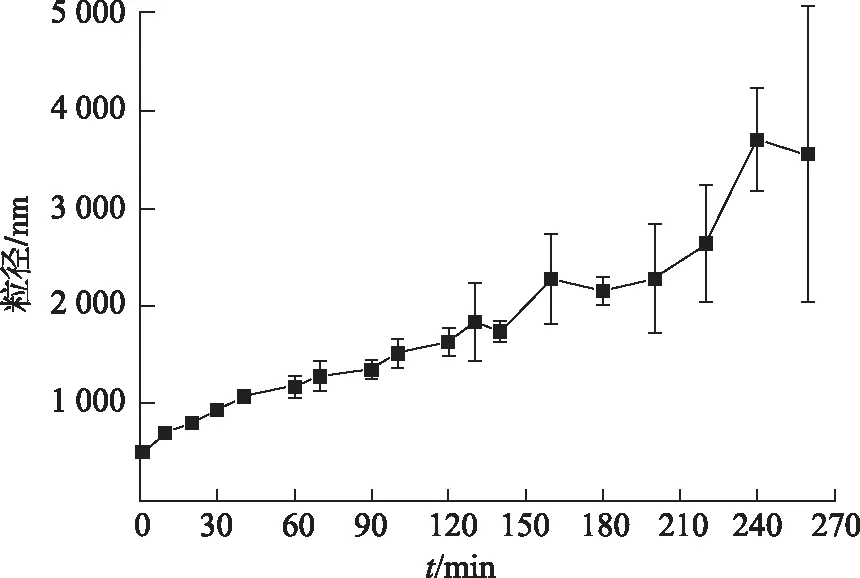
图5 粒径随时间变化曲线(n=3)
3 讨论
在利用固体分散技术提高难溶性药物溶解度的研究过程中,常常因为过饱和溶液中第二分散相的产生而使药物的溶解情况变得复杂。为更好地理解过饱和溶液中的第二分散相,如分散相的粒径、结晶时间,笔者在本研究通过溶剂转移法制备过饱和溶液,考察了在没有辅料存在的情况下,利托那韦药物的过饱和溶液发生相分离、产生第二分散相的临界浓度,进一步考察了在该分离浓度下第二分散相的结晶趋势和粒径变化。经过紫外消光、动态光散射、芘荧光探针法表征,发现利托那韦过饱和溶液发生相分离的浓度在大约37 μg·mL-1,相分离后形成可溶解的富药液滴,即第二分散相,随后该富药液滴开始结晶。另外,药物相分离浓度与其无定型状态溶解度有关。上述实验结果与文献研究结果基本一致[15]。偏光显微镜下的观察结果再次验证了上述结论。需要注意的是,并非所有过饱和溶液都可以发生相分离形成第二分散相,第二分散相的形成取决于溶液中的药物浓度是否超过了该药物无定型的溶解度[6]。同时,由于各种辅料的性能差异,药物-聚合物-水之间相互作用差异,导致不同辅料存在时,药物的溶出速率有较大差异,药物溶出后存在状态亦是多样化的[12,18-19],辅料或制备工艺对第二分散相的粒径增长控制越好或保持不结晶的时间越长,可能对提高生物利用度越有效果。
笔者在本研究系统全面地研究了利托那韦过饱和溶液中第二分散相的形成和特性,这对需要知道最大可达到的游离药物浓度的情况具有重要意义,例如在药物输送中用于构建剂量反应曲线,以及在确定最大生物接触限值时。另外,通过测定形成第二分散相时的浓度,对该体系中相边界的位置和形状进行更定量的了解,尤其是它们如何随化合物性质、离子强度、温度、辅料和浓度产生方法的变化而变化[15]。通常,辅料会影响第二分散相粒径、稳定性和结晶行为,为后续探究辅料和制备工艺对过饱和体系中第二分散相的影响,筛选具有优异结晶抑制特性的聚合物和制备工艺提供研究基础,并有助于指导后续新型药用辅料的开发。普渡大学Taylor课题组研究表明,带电荷的辅料降低了第二分散相聚结的动力学,但是对结晶动力学具有可变的影响,促进或抑制了结晶,通过适当选择配方成分,可以促进第二分散相胶体液滴的形成,并抑制其聚结和结晶[16]。此外,过饱和体系越来越多地应用于药物递送系统,固体分散体就是一个典型的过饱和给药体系,像自乳化药物传递系统、纳米制剂等也是基于药物处于过饱和状态,进而提高生物利用度的制剂技术。通过对过饱和体系的理解和认识,有助于合理设计过饱和药物传递系统,并对难溶性药物进行更深入的探索和利用。
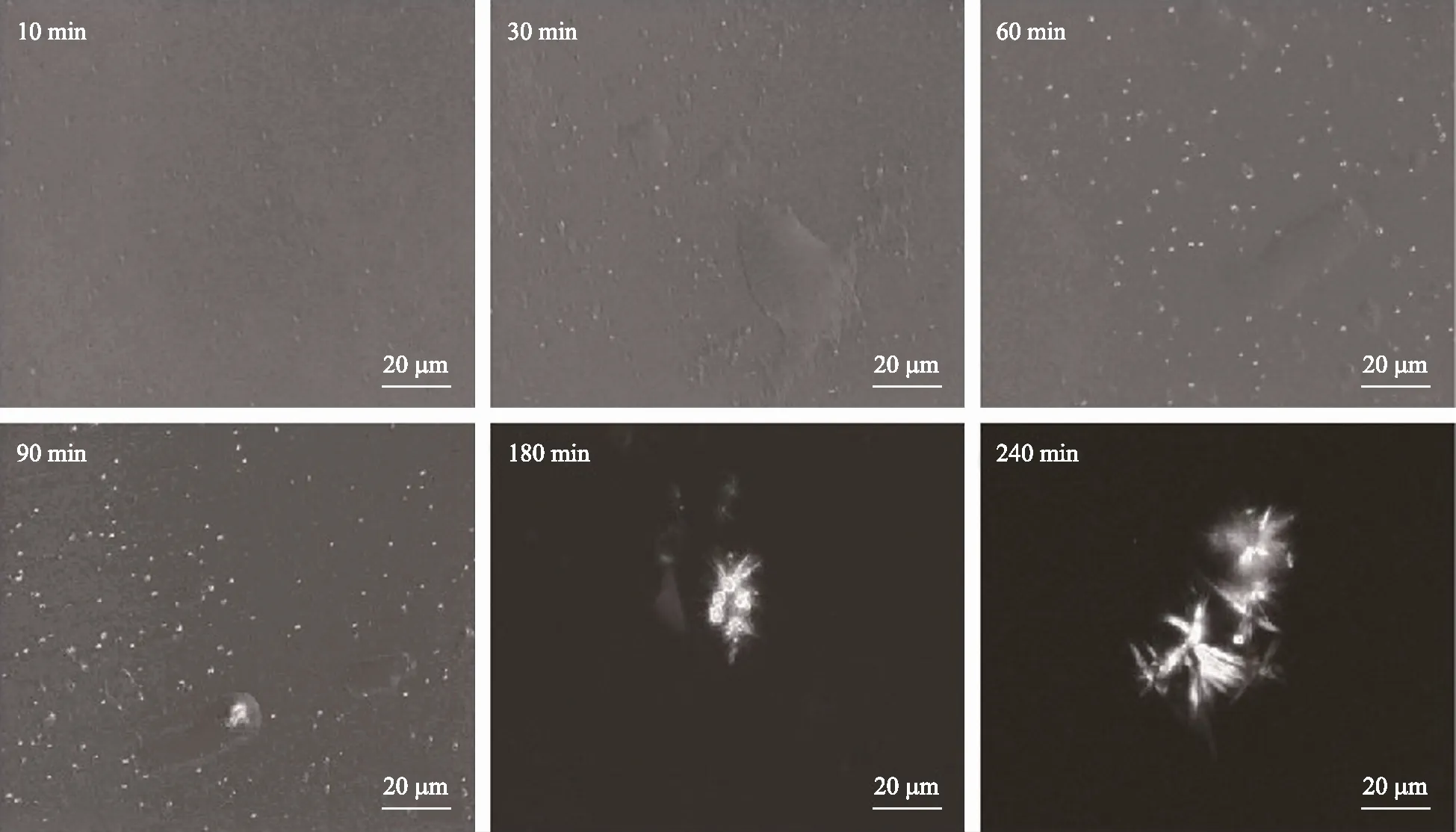
图6 过饱和溶液在不同时间点的偏光照片
