锌指蛋白512B基因变异的肌萎缩侧索硬化1例报告
2018-09-13幸伟芳洪铭范
幸伟芳 洪铭范
广东药科大学附属第一医院神经内科,广东 广州 510000
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)是运动神经元病中最常见的类型,以进行性加重的骨骼肌无力、萎缩、肌束颤动、延髓麻痹和锥体束征为主要临床表现。随着分子生物技术的飞速发展,对该病的研究取得了突破性的进展。在FALS的遗传学研究中,许多诱发ALS发病的易感基因已被确定,11种基因可以描述FALS的致病突变,包括SOD1、ALS2、VAPB、SETX、ANG、DCTN1、TARDBP、NEFH、FUS、DAO和OPTN。关于SALS的易感基因已有数十篇来自候选基因的相关研究报道,包括NEFH、ANG、APEX、HFE、PON、SMN1、SMN2和VEGF[1],对于SALS的基因研究极为重要。本文报道1例新的热点基因—锌指蛋白512B(ZNF512B)基因合并DES基因变异的肌萎缩侧索硬化病例。
1 病历摘要
患者男,58岁。因“双上肢无力10月余,加重伴肌肉萎缩3个月”入我院。患者于10个月前无明显诱因出现左上肢无力,稍欠灵活,约3个月后出现右上肢无力,可抬起,不影响日常生活,当时未予重视;3个月前患者双上肢无力加重,可抬起,持物尚稳,开始出现全身多处肌肉萎缩,双手手指笨拙,不能完成穿、脱衣服;1月前患者病情进一步加重,双上肢勉强可抬起,不能持物,肌肉萎缩明显,随后伴有饮水呛咳,声音变小,构音欠清,自觉抬颈困难,并出现双下肢乏力感,行走尚稳,无吞咽困难、无气促和呼吸费力。起病以来体质量下降约30 kg。
体格检查:言语欠清,行走尚稳,抬颈无力,双手部大小鱼际肌、骨间肌、蚓状肌、前臂、上臂、肩胛带肌群明显肌萎缩,双下肢腓肠肌明显肌萎缩,刺激受累部位肌群时有明显肌束震颤;双上肢肌力约3级,双下肢肌力5-级;双上肢肌张力正常,双下肢肌张力中度增高;指鼻试验不能完成;双下肢膝腱、跟腱反射亢进;左侧Hoffmann征阳性。颈3、4平面针刺觉过敏,右上肢针刺觉稍减退;余体格检查未见明显异常。
辅助检查:神经传导速度及肌电图提示:双侧上肢以运动支轴索性损害为主,部分感觉轻度受累,无传导阻滞,双侧下肢以感觉受累为主(患者有糖尿病病史),运动支无明显受累;上肢、下肢、胸椎旁肌、腰椎旁肌、胸锁乳突肌神经源性损害;右侧尺神经低频刺激阳性提示病情进展较快。肌电图诊断:神经电生理检查提示广泛的运动神经元损害。头颅CT示:(1)右侧顶枕叶交界处脑梗死;(2)右侧基底节区腔隙性梗死;(3)脑萎缩;(4)右侧上颌窦炎症并黏膜下囊肿。颈胸椎MRI平扫:颈胸椎退行性变,C 3/4~T 7/胸1椎间盘中央型突出,以C 5/6椎间盘向后突出为著,C 4/5、C 5/6椎间盘层面颈髓变性;胸椎未见明显异常。脑电图正常。胸片正常。正常范围心电图。腰穿:压力为200 mmH2O,脑脊液清亮,脑脊液常规:潘氏球蛋白定性 阳性(+),白细胞计数(体液)0(106/L);脑脊液生化:微量总蛋白 1.01 g/L;脑脊液涂片找细菌、抗酸杆菌、找隐球菌阴性。血脂全套(含Lpa/Hcy):低密度脂蛋白3.10 mmol/L,甘油三酯 2.06 mmol/L;血常规、电解质、肾功能、肝功能、心肌酶、心肌损伤标志物、脑钠肽、凝血四项、红细胞沉降率、免疫四项、风湿四项、空腹血糖、餐后2 h血糖、糖化血红蛋白、尿常规、大便常规+潜血均未见异常。遗传病医学外显子组基因测序见图1~3。
2 讨论
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)的诊断主要基于患者临床表现、世界神经病学联盟公布的共识,同时排除临床表现相似的疾病。根据改良Airlie House(含Awaji-Shima)诊断标准:(1)确诊ALS:由临床或电生理学证据证实,存在延髓和≥2个脊髓区域的上、下运动神经元损害证据,或存在3个脊髓区域的上、下运动神经元损害证据。(2)很可能ALS:由临床或电生理学证据证实,存在≥2个区域的上、下运动神经元损害证据,且某些上运动神经元体征在下运动神经元证据的上部。(3)可能ALS:由临床或电生理学证据证实,1个区域上、下运动神经元功能障碍,或≥2个区域孤立的上运动神经元损害体征,或某些下运动神经元体征在上运动神经元体征的上部。根据患者的病情发展、临床体格检查和神经电生理检查,可临床确诊为:“肌萎缩侧索硬化(ALS)”。
ZNF512B基因最初是在KAZUSA Human cDNA的基因工程研究中作为KIAA1196被标识的[2],包含6个C2H2型锌指结构域,编码一种含有893个氨基酸的蛋白质,是参与细胞分化、胚胎发育等的转录因子。ZNF512B在许多组织中均表达,包括脑和脊髓[3]。为了研究ZNF512B基因与ALS的关系,IIDA等[4]对一大批日本ALS患者的基因数据库进行了单核苷酸多态性(single nucleotide polymorphisms,SNPs)分析,结果通过基因图谱筛选发现了一个功能性SNP片段与ALS显著相关。rs2275294位于ZNF512B基因的12内含子上,含有rs2275294的基因组区域可以作为ZNF512B启动子的增强子,包含3种等位基因:CC、CT和TT,其中CC和CT属于ALS的易感性等位基因,该等位基因削弱了ZNF512B启动子的增强活性,并减弱其对于核蛋白质的结合能力,结果导致ZNF512B表达水平较低,从而导致ALS患病风险增加[4]。存在新型风险等位基因的ALS患者中,ZNF512B基因表达减少、血清和血浆中TGF-β水平的减少,从而导致神经元保护作用减弱。具有该风险等位基因的个体,其患病风险增高、病程进展迅速、临床预后较差。对ZNF512B调控TGF-β在ALS发病机制中所起作用的探索,将有助于开发针对ALS治疗的新对策,引人注目的是,该领域研究目前仅在日本人群中有相关报道。综上,通过对患者进行遗传病医学外显子组基因测序,亦支持“肌萎缩侧索硬化(ALS)”该诊断。
DES基因如发生致病性变异可引起肌原纤维肌病I型,以常染色体显性或常染色体隐性的方式遗传。肌原纤维肌病是一组病理上以异常蛋白沉积为主要特点的遗传异质性肌肉疾病,目前已知肌原纤维病的致病基因包括结蛋白(DES)[5]、αB-晶体蛋白(CRYAB)[6]、Z盘选择性剪接PDZ蛋白(ZASP)[7]、肌收缩蛋白(MYOT)[8]、细丝蛋白(FLNC)[9]等。主要临床特征表现为先出现远端肌无力,接着出现近端肌无力,影响肌萎缩等肌病症状、扩张型心肌病、肥厚型心肌病等心肌病症状。通常发病年龄在20~30岁。此外,该基因如发生致病性变异还可引起扩张型心肌病II型、神经性肩胛腓骨肌综合征Kaeser型、肌肉萎缩症2R型。肌原纤维肌病诊断主要依靠病理检查。遗憾的是,本病例中未对患者进行肌肉病理检查,尚未明确患者有无合并肌原纤维肌病可能。
查阅相关文献可发现目前国内尚未有人报道过ZNF512B基因变异的ALS病例。本文报道1例锌指蛋白512B(ZNF512B)基因合并DES基因变异的肌萎缩侧索硬化病例,希望能对未来SALS遗传学研究中起到一定帮助。
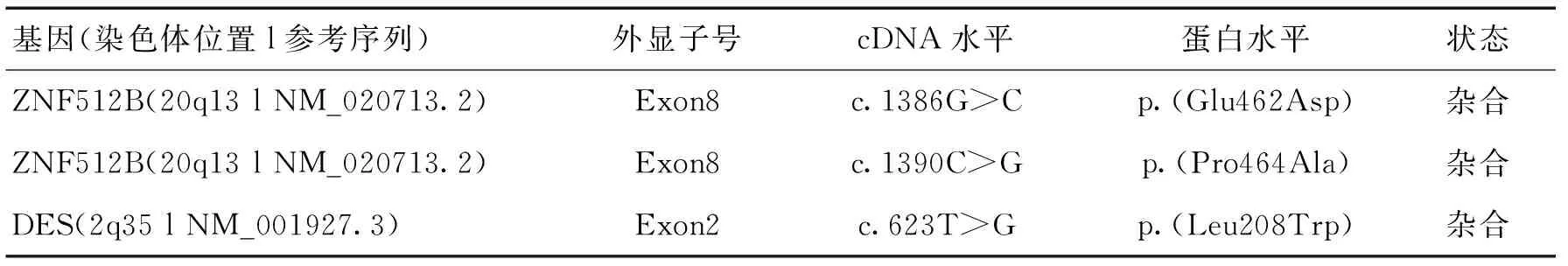
表1 总结图1~3内容分析

图1 遗传病医学外显子组(Exon8)基因测序Figure 1 Genetic disease medical exome (Exon8) gene sequencing
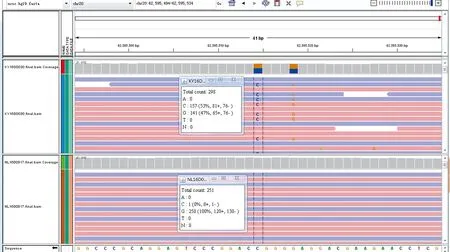
图2 遗传病医学外显子组(Exon8)基因测序Figure 2 Genetics disease exome (Exon8) gene sequencing

图3 遗传病医学外显子组(Exon2)基因测序Figure 3 Genetics disease exome (Exon2) gene sequencing
