伴肌萎缩重症肌无力4例分析并文献复习
2018-05-30雷国华田娜娜马艳艳周金津李尊波熊葶刘建军党根喜
雷国华 田娜娜 马艳艳 周金津 李尊波 熊葶 刘建军 党根喜
重症肌无力(myasthenia gravis,MG)是神经-肌肉接头(nerve-muscle joints,NMJ)传递障碍性疾病,由于自身抗体结合于NMJ突触后膜组分,主要是结合乙酰胆碱酯酶受体(acetylcholine receptor,AChR)而导致的器官特异性自身免疫性疾病[1]。MG的年发病率为(0.3~2.8)/10万[2],伴肌萎缩的MG更为罕见。现对作者医院2003—2016年诊治的4例伴肌萎缩MG患者的临床资料进行总结,并结合文献对其临床特点、抗体及电生理等特征进行讨论,以提高临床对该病的认识。
1 临床资料
1.1一般情况收集2003—2016年作者医院神经内科收治MG患者108例,其中4例(3.7%)患者伴肌萎缩。女3例、男1例,年龄依次为52、68、49、36岁,发病年龄依次为51、54、44、26岁。均以眼睑下垂或复视症状起病,其中病例1起病时被误诊为“双侧外展神经麻痹”。进展为全身型MG的时间依次为12、168、52、120个月。发生肌萎缩前4例患者的改良0sserman分型为ⅡB型1例,Ⅲ型1例,Ⅳ型2例;发生肌萎缩的病程依次为1.0、14.0、4.3、10.0年,肌萎缩发生部位为舌肌2例(图1A、B),舌肌+面肌萎缩1例(图1C、D),手肌萎缩1例(图1E)。入院时其中3例患者均表现为眼睑下垂、吞咽困难、饮水呛咳,4例患者肌无力均表现为晨轻暮重、活动后加重、休息后减轻等特点。4例患者新斯的明试验均阳性。胸部CT检查结果显示,2例存在胸腺瘤(病例2、3),1例胸腺增生(病例4),1例未见异常(病例1),其中病例3行胸腺切除术,术后病理示B2型胸腺瘤。病例4肌酸激酶值239 U/L。4例患者具体临床特征及治疗方案见表1。
1.2抗体检查采用放射免疫法检测自身抗体,其中1例肌肉特异性酪氨酸激酶抗体(muscle-specific receptor tyrosine kinase,MuSK-Ab)阳性,另3例均为AChR-Ab阳性(表1)。
1.3神经电生理检查4例MG患者重复神经刺激(RNS)检测均表现为低频刺激波幅递减,受累面神经6条、副神经3条、尺神经2条,低频刺激(3 Hz、5 Hz)面神经波幅递减11%~35%,副神经波幅递减17%~39%,尺神经波幅递减12%~16%。其中病例2右伸指总肌单纤维肌电图平均颤抖(jitter)增宽,达100 μs。病例4伴手肌萎缩的患者双上肢神经运动及感觉神经传导速度均在正常范围;针极肌电图检查结果显示,左侧外展拇短肌、左第一骨间肌及左侧外展小指肌针极肌电图静息及挪动针极时可见纤颤、正相电位,左侧外展小指肌及左第一骨间肌轻收缩运动单位电位时限增宽、电压增高,重收缩呈混合相、单纯-混合相,呈神经源性损害。
1.4病情进展及预后病例1和病例2病程中均出现2次肌无力危象,病例1和病例3均使用利妥昔单抗治疗后,病情稳定,病例2于2016-11出现肾病综合征,最终因多器官功能衰竭而死亡;病例4不同意进一步检查治疗而自动出院,未能行进一步检查以及追踪随访。其中病例1病情反复,2003-2016年期间共住院15次,多次给予大剂量甲泼尼龙冲击、静脉注射用丙种球蛋白冲击,免疫抑制剂先后换用环孢素、环磷酰胺、吗替麦考酚酯;2015-6-12因肺部感染诱发肌无力危象,经呼吸机辅助通气15 d后脱机,并给予“静脉注射用丙种球蛋白冲击+吗替麦考酚酯”治疗后,病情稳定;2015-9-23再次出现肌无力危象,呼吸机辅助治疗通气7 d后脱机,给予“大剂量甲泼尼龙冲击+静脉注射用丙种球蛋白+吗替麦考酚酯”治疗病情稳定后,2次查淋巴细胞亚群分析(流式细胞仪):CD19+CD20+细胞比例分别为8.4%及14.31%,于2015-10-21开始给予“利妥昔单抗”治疗,每周1次,连用4次,具体用法如下:治疗前30 min内给予“地塞米松注射液15 mg静脉注射+盐酸异丙嗪注射液25 g肌肉注射+酚麻美敏片372 mg口服”+“0.9%氯化钠注射液100 mL+利妥昔单抗注射液100 mg,静脉泵泵入,50 mL/h”,复查CD19+CD20+细胞比例为0%,此后病情稳定。
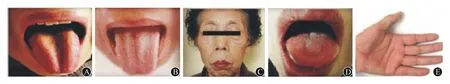
注:MG:重症肌无力,表1同;A、B:分别为病例1、3伴舌肌萎缩;C、D:病例2伴面肌及舌肌萎缩;E:病例4伴手肌萎缩 图 1 4例MG患者伴肌萎缩部位
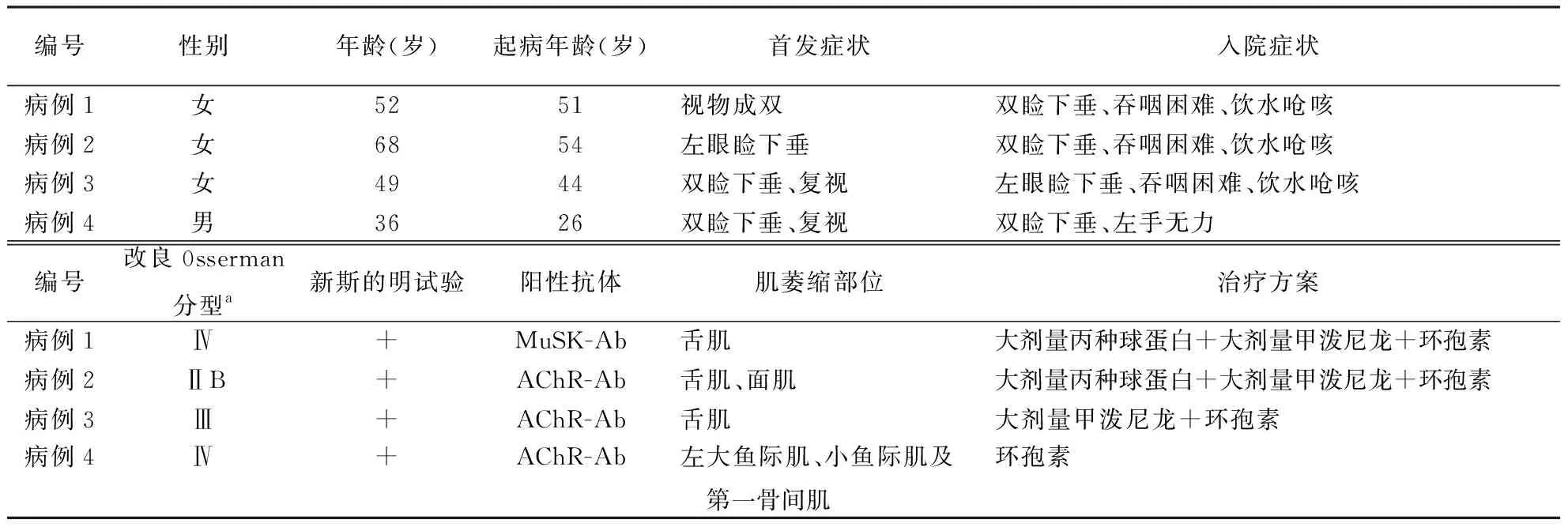
编号性别年龄(岁)起病年龄(岁)首发症状入院症状病例1女5251视物成双双睑下垂、吞咽困难、饮水呛咳病例2女6854左眼睑下垂双睑下垂、吞咽困难、饮水呛咳病例3女4944双睑下垂、复视左眼睑下垂、吞咽困难、饮水呛咳病例4男3626双睑下垂、复视双睑下垂、左手无力编号改良0sserman分型a新斯的明试验阳性抗体肌萎缩部位治疗方案病例1Ⅳ+MuSK-Ab舌肌大剂量丙种球蛋白+大剂量甲泼尼龙+环孢素病例2ⅡB+AChR-Ab舌肌、面肌大剂量丙种球蛋白+大剂量甲泼尼龙+环孢素病例3Ⅲ+AChR-Ab舌肌大剂量甲泼尼龙+环孢素病例4Ⅳ+AChR-Ab左大鱼际肌、小鱼际肌及 第一骨间肌环孢素
注:a为发生肌萎缩前的分型;MuSK-Ab:肌肉特异性酪氨酸激酶抗体;AChR-Ab:乙酰胆碱酯酶受体抗体
2 讨论
2.1伴肌萎缩MG罕见MG是临床中较少见的疾病,De Assis等[3]对752例MG患者进行研究发现仅10例(1.3%)出现肌萎缩。本文4例(3.7%)出现肌萎缩,其中3例为舌肌萎缩,1例为手肌(左侧大鱼际肌、小鱼际肌及第一骨间肌)萎缩。
2.2抗体与伴肌萎缩MGMartignago等[4]通过MG诊断标准、临床症状及RNS筛选出6例MuSK-Ab阳性MG和7例AChR-Ab阳性MG患者进行对比研究,结果发现两组中均存在有肉眼可见的舌肌萎缩,但MuSK-Ab阳性MG出现舌肌萎缩的频率更高;进一步研究发现,上述两组MG患者有不同的组织病理学模式,AChR-Ab阳性MG患者的骨骼肌活检可见Ⅰ型和Ⅱ型肌纤维萎缩因子较高,对四肢肌的影响较重,而MuSK-Ab的局部作用使MuSK-Ab阳性MG患者发生骨骼肌萎缩较少见,多为延髓肌萎缩。本组4例患者中,2例AChR-Ab阳性MG出现舌肌萎缩,可见虽然MuSK-Ab阳性MG出现舌肌萎缩的频率高,但AChR-Ab阳性MG患者也可出现舌肌萎缩。Cenacchi等[5]研究发现MuSK-Ab阳性MG活检组织的超微结构提示严重的肌病改变,如肌原纤维丧失,线粒体肿胀和嵴(线粒体向内突出的皱襞)破裂,而AChR-Ab阳性MG最常见的是肌纤维萎缩,肌原纤维排列紊乱。Samuraki等[6]发现MG肌萎缩可由运动终板的肌纤维失神经支配引起,也可因终板和突触膜异常终端扩张引起。
2.3电生理与肌萎缩RNS检测是诊断NMJ病变的主要电生理学手段,国外资料显示在MG中RNS检查结果阳性率为41%~95%[7]。陈玉萍等[8]分析436例患者低频RNS检查阳性率达73.85%。虽然本文仅报道4例患者,但低频RNS结果均为阳性表现。崔丽英等[9]对398例MG患者进行低频RNS检查发现腋神经刺激的低频RNS阳性率最高,其次是面神经,尺神经刺激最低。本文报道的4例患者,面、副神经刺激递减幅度明显,尺神经刺激递减幅度低。
2.4MG神经源性损害国外文献报道,合并肌源性损害的MG发生率为10.0%~37.5%[10],而合并神经源性损害的MG国内外报道较少见。神经源性损害的肌电图表现除可见失神经电位外,主要表现为宽时限、高波幅的运动单位动作电位,病例4患者肌电图表现为神经源性损害。Samuraki等[6]报道了1例神经源性损害的MG患者,为28岁日本男子,以复视和左上肢无力起病,6个月后出现双上肢无力及双睑下垂,症状与病例4类似,但该患者无胸腺瘤,肌酸激酶正常。de Pasqua等[11]报道了671例肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)患者中,5例患者为由MG逐渐发展为ALS,2例以眼部症状起病,3例以肢体无力起病。Gotaas等[12]和Tzartos等[13]认为LRP4抗体在运动神经元和NMJ处的作用至关重要,两种疾病之间可能是通过调节性T淋巴细胞的免疫调节相联系[14-15]。伴神经源性损害的MG是否合并ALS需要进一步研究。
2.5利妥昔单抗治疗MG利妥昔单抗是针对B细胞表面CD20抗原产生作用[16]。 Hehir等[17]研究了利妥昔单抗治疗MuSK-Ab阳性MG患者24例,对照组31例,结果提示利妥昔单抗对MuSK-Ab阳性MG更有效。本文2例患者在病程进展中经利妥昔单抗治疗后,病情趋于稳定。
综上所述,伴肌萎缩MG患者较为罕见,MuSK-Ab及AChR-Ab阳性MG患者均可出现肌肉萎缩,其机制有待进一步明确,早期行神经电生理检查、AChR-Ab及 MuSK-Ab测定有助于明确诊断。利妥昔单抗治疗MuSK-Ab阳性MG有助于患者病情趋于稳定。伴神经源性损害MG是否合并ALS值得关注。
[1]Sanders DB,Wolfe GI,Benatar M,et al.International consensus guidance for management of myasthenia gravis[J]. Neurology,2016,87(4):419-425.
[2]Deenen JC,Horlings CG,Verschuuren JJ,et al.The epidemiology of neuromuscular disorders: A comprehensive overview of the literature[J]. J Neuromuscul Dis,2015,2(1):73-85.
[3]De Assis JL,Marchiori PE,Scaff M.Atrophy of the tongue with persistent articulation disorder in myasthenia gravis: report of 10 patients[J]. Auris Nasus Larynx,1994,21(4):215-218.
[4]Martignago S,Fanin M,Albertini E,et al.Muscle histopathology in myasthenia gravis with antibodies against MuSK and AChR[J]. Neuropathol Appl Neurobiol,2009,35(1):103-110.
[5]Cenacchi G,Papa V,Fanin M,et al.Comparison of muscle ultrastructure in myasthenia gravis with anti-MuSK and anti-AChR antibodies[J]. J Neurol,2011,258(5):746-752.
[6]Samuraki M,Furui E,Komai K,et al.Myasthenia gravis presenting with unusual neurogenic muscle atrophy[J]. Muscle Nerve,2007,36(3):394-399.
[7]Conti-Fine BM,Milani M,Kaminski HJ.Myasthenia gravis: past,present,and future[J]. J Clin Invest,2006,116(11):2843-2854.
[8]陈玉萍,王卫,魏东宁.低频重复电刺激检查在重症肌无力诊断中的临床意义[J]. 中华医学杂志,2011,91(17):1178-1180.
[9]崔丽英,刘明生.重症肌无力的电生理诊断[J]. 中国实用内科杂志,2006,26(04):249-251.
[10]Odabasi Z,Kuruoglu R,Oh SJ.Turns-amplitude analysis and motor unit potential analysis in myasthenia gravis[J]. Acta Neurol Scand,2000,101(5):315-320.
[11]de Pasqua S,Cavallieri F,D′ Angelo R,et al.Amyotrophic lateral sclerosis and myasthenia gravis: association or chance occurrence?[J]. Neurol Sci,2017,38(3):441-444.
[12]Gotaas HT,Skeie GO,Gilhus NE.Myasthenia gravis and amyotrophic lateral sclerosis: A pathogenic overlap[J]. Neuromuscul Disord,2016,26(6):337-341.
[13]Tzartos JS,Zisimopoulou P,Rentzos M,et al.LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients[J]. Ann Clin Transl Neurol,2014,1(2):80-87.
[14]Berrih-Aknin S,Le Panse R.Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms[J]. J Autoimmun,2014,52:90-100.
[15]Henkel JS,Beers DR,Wen S,et al.Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival[J]. EMBO Mol Med,2013,5(1):64-79.
[16]Emer JJ,Claire W.Rituximab: a review of dermatological applications[J]. J Clin Aesthet Dermatol,2009,2(5):29-37.
[17]Hehir MK,Hobson-Webb LD,Benatar M,et al.Rituximab as treatment for anti-MuSK myasthenia gravis[J]. Neurol,2017,89(10):1069-1077.
