2,3,4,6-四-O-苄基-吡喃葡萄糖合成工艺改进
2017-09-03曹凌峰姜国平季铭键朱锡忠
曹凌峰,姜国平,季铭键,朱锡忠
(浙江合糖科技有限公司,浙江金华321016)
精细化工
2,3,4,6-四-O-苄基-吡喃葡萄糖合成工艺改进
曹凌峰,姜国平,季铭键,朱锡忠
(浙江合糖科技有限公司,浙江金华321016)
以甲基葡萄糖苷为起始原料,经改进的Williamson苄醚化反应,选择性水解反应得到目标产物。优化了苄醚化及选择性水解反应条件。苄醚化以甲基葡萄糖苷、氢氧化钠、氯化苄为原料,以正辛烷为带水剂,125℃~130℃保温回流脱水反应8 h,产率为98.0%~99.0%。水解反应以丙酸替代乙酸为溶剂,高氯酸为催化剂,85℃~90℃保温回流脱水反应8 h,产率为65.0%~70.0%。各步产物结构经FTIR、HPLC-MASS、1H NMR、13C NMR确证。
2,3,4,6-四-O-苄基-吡喃葡萄糖;Williamson醚化法;碱金属氢氧化物;带水剂;选择性水解
2,3,4,6-四-O-苄基-D-吡喃葡萄糖(简称四苄基葡萄糖,TBG),是一种白色至类白色粉末或结晶,作为手性源,是多种药物和生物活性物质合成的重要中间体,具有多种用途。四苄基葡萄糖是合成治疗糖尿病药物伏格列波糖的关键中间体[1]。四苄基葡萄糖也可用于拜耳公司开发的抗Ⅱ型糖尿病药物。四苄基葡萄糖与三氯乙腈反应[3],可用于合成四苄基葡萄糖基三氯乙酰亚胺酯,该产物是非常重要的糖基给予体。
四苄基葡萄糖合成方法已有较多研究与报道。焦岩等[4]以蔗糖为起始原料,先与醋酐反应将其制成蔗糖八醋酸酯,再苄基化得八苄基蔗糖,经水解制得四苄基葡萄糖和四苄基果糖的混合物,再经结晶和柱分离分别得到四苄基葡萄糖和四苄基果糖,水解收率29%,三步反应总收率为23.5%,收率较低。烯丙苷法[5]、n-戊-4-烯苷[6]法水解收率较高,分别为65%和85%,但原料烯丙醇(或溴丙烯)高毒,而4-戊烯-1-醇价格昂贵,故也不适合作为工业化制备四苄基葡萄糖的方法。另有报道[7],以甲基葡萄糖苷(MeG)为原料,以氢化钠为碱,进行苄醚化反应;再经选择性水解制得四苄基葡萄糖[3]。该法优点是,甲基葡萄糖苷来源广泛,价格便宜,可采用淀粉、纤维素及葡萄糖为原料制得。不足之处是:氢化钠易燃易爆且为石蜡油分散体,存在安全与质量风险,不宜工业化。本实验室[8]采用了以碱金属氢氧化物为碱,在带水剂存在下,把甲基葡萄糖苷进行苄醚化反应取得良好效果。另外,文献报道的水解反应多采用醋酸—水体系,收率不高,约45%~50%。
作者在实验室已有的工作基础上,改进苄醚合成与选择性水解工艺。进一步优化以碱金属氢氧化物为碱的苄醚化工艺,以甲基葡萄糖苷为原料,氢氧化钠为碱,在正辛烷中回流带水条件下与氯化苄反应,制得甲基四苄基葡萄糖苷(MeTBG)。以丙酸-水溶液代替醋酸-水溶液为水解反应体系,利用其对甲基四苄基葡萄糖苷和产物四苄基葡萄糖的溶解度差异,提高水解选择性,将水解一次收率提高至65.0%~70.0%。反应方程式如下所示:
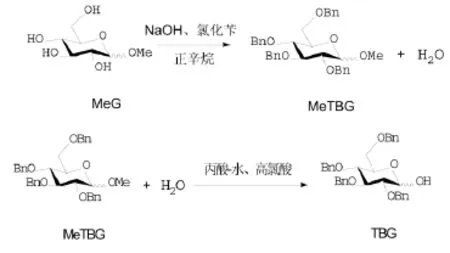
1 实验部分
1.1 试剂与仪器
甲基葡萄糖苷,质量分数≥99.0%,北京杨村化工有限责任公司;正辛烷、氯化苄、氢氧化钠、高氯酸、硫酸、对甲苯磺酸、2-己酮等均为CP,国药集团试剂有限公司。
HPLC:Waters 515 HPLC Pump,Waters2414示差检测器(美国Waters公司),色谱柱TSKgel G2000H(日本TOSOH公司);NEX-US670型傅立叶变换红外光谱仪(KBr压片),美国NICOLET公司;AV600型核磁共振波谱仪(DCCl3为溶剂,TMS为内标),德国BRUKER公司;液质联用:LC-30A型UPLC仪(日本岛津公司),TripleTOF4600质谱仪(美国AB Sciex公司)。
1.2 甲基四苄基葡萄糖苷的制备
在装有回流分水器的250 mL三颈瓶中依次加入9.71 g甲基葡萄糖苷,9.2 g氢氧化钠,60 mL正辛烷,搅拌下升温回流带水反应至基本无水蒸出。滴加30 g氯化苄,后继续保温回流带水反应,HPLC监测反应进程,约需时8 h。过滤除去固体物质。滤液减压浓缩至干,得金黄色黏稠状液体30.44 g,HPLC含量89.4%,收率98.2%。所得甲基四苄基葡萄糖苷无需精制即可用于水解反应。1H NMR(DCCl3,600 MHz),δ:7.60~6.91(m,20H,4×C6H5),4.98(d,J=10.8 Hz,1H),4.87~4.75(m,3H),4.70~4.50(m,3H),4.50~4.40(m,2H),3.98(t,J=9.3 Hz,1H),3.78~3.68(m,2H),3.63(dt,J=9.4,4.8 Hz,2H),3.56(dd,J=9.6,3.5 Hz,1H),3.37(s,3H);13CNMR(CDCl3,ppm)δ:138.86,138.32,138.22,137.97,129.79,128.53,128.50,128.45,128.44,128.41, 128.40,128.28,128.19,128.13,128.02,127.96,127.90,127.74,127.71,127.68,127.64,98.27,82.19,79.89,77.72,77.35,77.14,76.92,75.81,75.09,73.54,73.45,70.11,68.53,55.23;IR(KBr)ν:3 062,3 028,1 496,1 452,2 900,1 359,1 043、1 026,732,694 cm-1;UPLC-MS,m/Z:555.26(M+H+),572.3(M+H2O),577.25(M+Na+)。
1.3 四苄基葡萄糖的制备
在带有回流装置的250 mL四颈瓶中依次加入述所得30.44 g甲基四苄基葡萄糖苷,60 g丙酸-水溶液(含水25%),1.78 g高氯酸(70%~72%),搅拌升温至85℃~90℃,保温反应8 h。保温反应毕,加入氢氧化钠猝灭反应。将溶剂蒸出,加入60 ml 2-己酮溶解,后水洗至中性。过滤,滤液冷却至10℃~15℃进行结晶,抽滤,得白色固体粉末,烘干,得18.5 g,HPLC含量99.3%收率69.3%。Mp 152.4℃~153.0℃。1H NMR(DCCl3,600 MHz),δ:7.43~7.04(m,20H,4×C6H5),5.21(d,J=2.8 Hz,1H),5.06~4.35(m,8H,4×CH2),4.12~3.91(m,2H),3.78~3.51(m,4H),3.41~ 3.37(m,1H);13C-NMR(CDCl3,ppm)δ:138.75,138.60,138.45,138.26,138.02,137.96,137.87,137.79,128.55,128.51,128.46,128.44,128.43,128.19,128.11,128.04,128.01,127.94,127.86,127.81,127.78,127.75,127.74,127.68,97.53,91.30,84.64,83.16,81.82,80.03,77.91,77.83,77.36,77.15,76.94,75.78,75.73,75.07,75.04,74.77,74.66,73.53,73.51,73.22,70.25,68.97,68.67;IR(KBr)ν:3 344.75,3 087.39,3 062.57,3 028.93,2 904.82,2 858.41,1 604,1 587,1 495.74,1 451.45,1 355.65,1 084.44,1 071.92,1 025.43,903.69,741.83,692.45 cm-1;UPLC-MS,m/Z:541.28(M+H+),558.28(M+H2O),563.24(M+Na+)。
2 结果与讨论
2.1 甲基四苄基葡萄糖苷制备条件优化
本实验室对williamson苄醚化反应进行了改进[8],成功应用于葡萄糖衍生物的苄醚化反应,取得较好效果。对不同底物反应溶剂、碱、苄醚化试剂、配比等因素已进行了考察。本文在已有工作基础上,进一步考察氯化苄加料方式、反应温度的影响。
2.1.1 氯化苄加料方式对苄醚化反应的影响
按1.2所述进行实验,改变氯化苄滴加方式,相同配比下,考察对反应的影响。结果如表1所示,所列为液相检测目标产物、中间产物及杂质含量。

表1 加料方式对转化率的影响Table 1 Effect of feeding mode on yield
由表1,根据脱出水量滴加氯化苄可提高原料利用率,减少中间产物,使反应完全且抑制副产物生成。
2.1.2 反应温度对苄醚化反应的影响
按1.2所述进行实验,改变反应温度,并抽真空,使得体系在实验温度下可回流带水,考察反应温度对苄醚化反应的影响。结果如表2所示,所列为液相检测目标产物、中间产物及杂质含量。
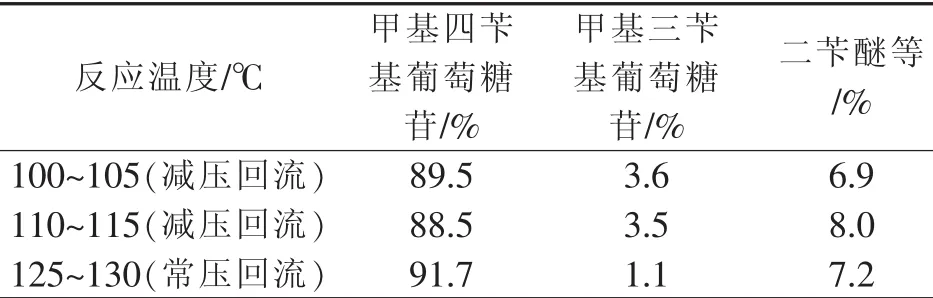
表2 反应温度对转化率的影响Table 2 Effectofreaction temperature on yield
由表2可知,125℃~130℃反应效果较好。反应温度高,醇钠化反应进行得更快,苄醚化反应也较完全。另外,采用减压回流分水方式更易使氯化苄逸出,损失原料,影响反应效果。
2.2 四苄基葡萄糖制备条件优化
甲基四苄基葡萄糖苷为黏稠性液体,水中溶解度低,因此采用合适的溶剂体系使底物、水、催化剂呈均相,将有利于反应进行。底物中的苄醚键虽比C1位甲苷键稳定,但在强酸、高温条件下仍不可避免地发生水解副反应。因此,水解反应条件应尽可能温和,以提高反应的选择性。此外,若可将产物从反应体系中及时移出,也可减少苄醚键水解,提高收率。本文采用丙酸-水代替文献的醋酸-水体系,考察了丙酸—水比例、催化剂、反应温度-时间等因素对反应的影响。
2.2.1 丙酸-水比例对水解反应收率的影响
按1.3所述进行实验,改变丙酸-水比例,相同配比下,考察对反应的影响。结果如表3所示。

表3 丙酸与水比例对水解反应收率影响Table 3 Effect ofpropionic acid/water ratio on yield
由表3可知,丙酸中含水20%~25%得到较高收率,超过25%时体系易分层,收率明显降低。研究发现,丙酸比醋酸对甲基四苄基葡萄糖苷有更好的溶解性能,反应温度下即使含水25%的丙酸水溶液(醋酸水溶液约为15%)仍能很好地溶解底物。反应体系含水量的增加也同时提高了水对底物的比例,推动反应正向进行。值得一提的是水比例增加至20%后,随反应进行,可观察到有白色固体析出,经分离鉴定为目标产物。目标产物在体系中析出,减少了其被进一步水解、破坏的几率。对比实验发现,在醋酸水体系中,仅能观察到少量絮状析出,大部分产物仍溶于体系,其苄醚基易被进一步水解。
2.2.2 催化剂对水解反应收率的影响
按1.3所述进行实验,催化剂类型,相同配比下,考察对反应的影响。结果如表4所示。

表4 催化剂对水解反应收率的影响Table 4 Effect of catalysts on yield
由上表可知,高氯酸的催化效果最好,这与上述几种酸的酸性强弱顺序一致。在有机酸-水体系中,氢离子离解被抑制,只有强酸才能有较好的离解,使氢正离子参与水解反应的催化过程。
2.2.3 反应温度、时间对水解反应收率的影响
本文研究了不同温度条件下的水解反应过程,并以液相色谱检测反应情况,以确定反应所需时间。结果如表5所示。
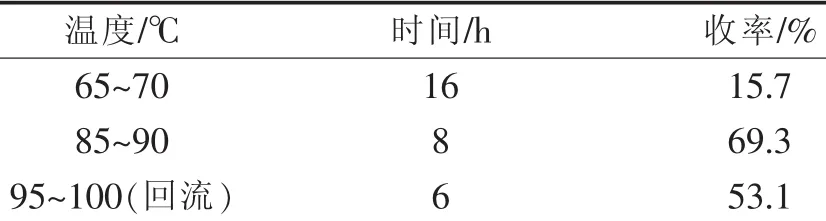
表5 反应温度对收率的影响Table 5 Effect of reaction temperature on yield
由表5可知,反应以85℃~90℃条件下水解8 h为佳。温度升高时反应选择性变差,致使收率降低;而过低的反应温度则反应速度大大降低,即使大幅度延长时间,其收率依然较低。
3 结论
(1)甲基葡萄糖苷、氢氧化钠、氯化苄在正辛烷中,回流分水条件下,125℃~130℃保温回流脱水反应8 h制得甲基四苄基葡萄糖苷,收率98%~99%。甲基四苄基葡萄糖苷无需进一步精制即可用于水解反应。以含水25%丙酸水溶液,高氯酸为催化剂,85℃~90℃保温反应8 h,经后处理制得四苄基葡萄糖,含量99.3%,收率69.3%。两步反应总收率67.8%~68.6%,较文献45%~50%有明显提高。经FTIR、UPLC-MASS、1H NMR对各产物结构进行表征,证实合成了目标产物。
(2)工业可行性较好。甲基四苄基葡萄糖苷制备过程中,避免了氢化钠的使用,氢氧化钠和氯化苄过量较少时即可反应完全,操作简单,环境友好具有良好的工业可行性。
(3)选择性水解反应制备四苄基葡萄糖过程中,充分利用原料与产物在水解体系中溶解度差异,调整丙酸-水比例,原料在体系中充分溶解的同时降低产物溶解度,在水解反应可顺利进行的前提下降将产物从体系中析出,降低了其被进一步水解转化为杂质的风险,最终提高了收率。
[1]Hiroshi F,Satoshi H.Synthesis of a branched-chain inosose derivative,a versatile synthon of N-substituted valiolamine derivatives from D-glucose[J].J.Org.Chem.,1992,57:3642-3650.
[2]张齐华,容元伟,高勇,等.米格列醇的合成[J].化学研究与应用,2014,26(7):1095-1098.
[3]许环军,康帅涛,李晓东,等.苄基保护的α-D-葡萄糖三氯乙酰亚胺酯的合成工艺及分离方法改进[J].沈阳药科大学学报,2012,29(5):352-354.
[4]焦岩,方志杰,江荣英.八苄基蔗糖及其水解反应产物的制备[J].化学试剂,2007,29(8):503-504,506.
[5]蔡孟深,李中军.糖化学基础、反应、合成、分离及结构[M].北京:化学工业出版社,2007:36.
[6]David R M,Vandana D,Bert Fraser-Reid B.n-Pentenyl glycosides permlt the chemospecific liberation of the anomeric center[J].J.Am.Chem.Soc,1988,110:5583.
[7]Jin X F,Meng X B.Facile synthesis of a core trisaccharide of laminin as a potential metastatic antagonist[J].Journal of Chinese Pharmaceutical Sciences,2008,17:272–276.
[8]姜国平,曹凌峰.改进的Williamson醚化法合成葡萄糖衍生物[J].精细化工,2017,34(1):87-91.
An Improved Method for Synthesis of 2,3,4,6-Tetra-O-benzyl-glucopyranose
CAO Ling-feng JIANG Guo-ping,JIMing-jian,ZHU Xi-zhong
(Zhejiang Synosetech Co.,Ltd.,Jinhua,Zhejiang 321016,China)
An improved method for synthesis of 2,3,4,6-tetra-O-benzyl-glucopyranose was reported. Using methyl-glucopyranoside as raw material,via improved Williamson etherification and selective hydrolysis,2,3,4,6-tetra-O-benzyl-glucopyranose was synthesized.The optimum conditions of Williamson etherification and selective hydrolysis were investigated.In Williamson etherification,using methyl-glucopyranoside,NaOH and benzyl chloride as raw material,using n-octane as water-carrying agent,refluxed at 125℃~130℃for 4.0 h,gave Methyl-2,3,4,6-tetra-O-benzyl-glucopyranoside in 98.2%yields.In selective hydrolysis,using propionic acid instead of common acidic acid as reaction solvent,using perchloric acid as catalyst,reaction at 85℃~90℃for 8.0 h,gave 2,3,4,6-tetra-O-benzyl-glucopyranose in 65.0%~70.0%. The structures ofcompounds obtained were confirmed by FTIR,UPLC-MASS,1H NMR.
2,3,4,6-tetra-O-benzyl-glucopyranose;Williamson etherification method;alkali metal hydroxide;water-carrying agent;selective hydrolysis
1006-4184(2017)7-0013-04
2017-01-21
金华市科学技术研究计划重点项目(批准号:2015-1-020)。
曹凌峰(1983-),男,浙江金华人,工程师,硕士,主要从事药物中间体合成工作。E-mail:clf@synose.com。
