一例遗传性凝血因子Ⅺ缺陷症患者表型诊断及基因分析
2017-06-21叶佳佳杨丽红郝秀萍陈必成
叶佳佳,杨丽红,郝秀萍,陈必成
(温州医科大学附属第一医院,浙江 温州 325015,1.检验科;2.外科实验室)
一例遗传性凝血因子Ⅺ缺陷症患者表型诊断及基因分析
叶佳佳1,杨丽红1,郝秀萍1,陈必成2
(温州医科大学附属第一医院,浙江 温州 325015,1.检验科;2.外科实验室)
目的:对1例遗传性凝血因子XI(FXI)缺陷症患者及其家系成员进行表型诊断和基因分析,探讨其发病的分子机制。方法:检测先证者及其家系成员(共3代8人)的血浆凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、纤维蛋白原含量(FIB)、凝血因子Ⅺ促凝活性(FXI∶C)及凝血因子Ⅺ抗原含量(FXI∶Ag)等指标以明确诊断;聚合酶链式反应(PCR)扩增FXI基因的所有外显子和侧翼序列,扩增产物纯化后直接测序,发现突变位点则反向测序予以证实;用PyMOL Viewer1.5.x软件构建野生型和突变型FXI蛋白模型,比对分析寻找突变前后空间构象及分子间作用力的变化。结果:先证者和其弟弟APTT、FXI∶C、FXI∶Ag均明显异常,分别为78.4 s、2.0%、6.8%和62.1 s、4.5%、10.0%;其家系成员的FXI∶C和FXI∶Ag均发现有不同程度下降。基因分析发现先证者和其弟弟FXI基因第6号外显子的g.15410G>A杂合无义突变导致Trp228stop及第12号外显子的g.25471C>G杂合错义突变导致Cys482Trp;其父亲、姐姐、女儿、外甥女均为Trp228stop的杂合子,而母亲、侄子则为Cys482Trp的杂合子。模型分析显示Cys482Trp突变并未破坏氨基酸间的天然氢键联系,但使该位点氨基酸与267位氨基酸的空间位阻变大,从而使蛋白的结构发生变化。结论:该遗传性FXI缺陷症患者FXI基因的Trp228stop无义突变和Cys482Trp错义突变与血浆FXI∶C及抗原水平降低有关。
遗传性凝血因子XI缺陷症;聚合酶链式反应;基因突变;模型分析
凝血因子XI(FXI),又称为血浆凝血活酶前质,主要由肝脏和巨核细胞产生。在传统凝血机制中,FXI在凝血因子X II与高分子量激肽原的作用下转变为活化的FXI(FXIa),它激活凝血因子IX,从而活化凝血级联反应。研究发现,FXI的主要作用是在血凝块形成后促进凝血酶的持续生成[1],而且FXIa还有间接抑制纤溶的作用,从而可以稳定已经形成的凝血块。遗传性FXI缺陷症是一种由于基因突变导致的遗传性疾病,呈常染色体隐性遗传,男女均可患病,在人群中的发病率为1/1 000 000到1/100 000,但在犹太人和法国巴斯克地区发病率较高[2]。患者出血表现轻微,甚至无出血症状,多数在纤溶活跃部位(口腔或泌尿生殖系)手术时因出现明显的凝血障碍而被诊断[3]。本研究通过对1例遗传性FXI缺陷症家系成员的表型检测及基因分析,初步探讨其分子致病机制,报告如下。
1 对象和方法
1.1 对象 先证者,女,32岁,因妊娠G2P1孕11周+3 d,阴道出血,临床诊断“先兆流产”,住院保守治疗。妊娠监督检查凝血常规发现活化部分凝血活酶时间(activated partial thromboplastin time,APTT)轻度延长(78.4 s),进一步检查发现凝血因子XI促凝活性(FXI∶C)和凝血因子XI抗原含量(FXI∶Ag)明显降低,分别为2.0%和6.8%,其他凝血指标均无明显异常,肝、肾功能正常。先证者自诉平时无自发出血或血栓等症状,但分娩时出血量较多,输入血浆后可纠正。征得先证者及其家系成员知情同意后进行后续实验,该家系遗传图谱见图1。150例正常健康体检者作为凝血指标对照,其中男78例,女72例,平均年龄35岁(17~56岁)。均无肝、肾功能疾病,无血栓或出血史。本研究经本院伦理委员会批准,所有受检者经知情同意后,抽取血样。
1.2 方法
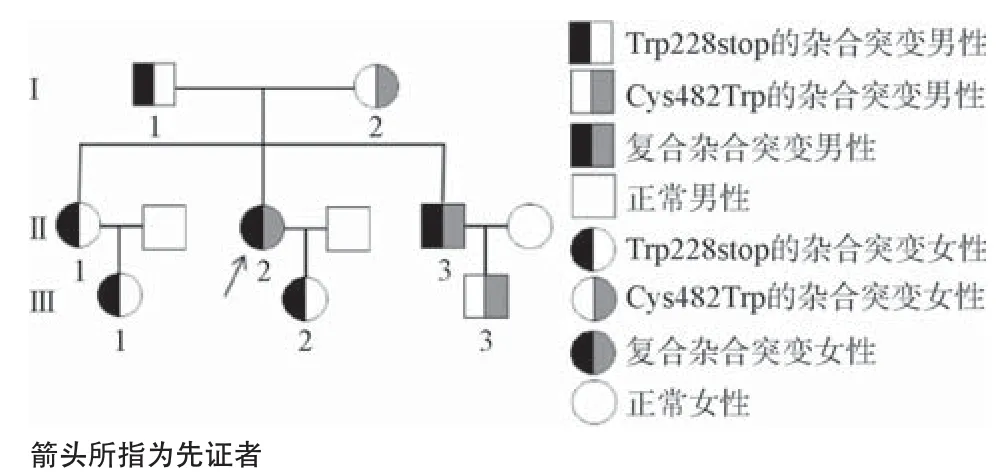
图1 遗传性FXI缺陷症先证者家系图
1.2.1 仪器和试剂:基因组DNA提取试剂盒及PCR扩增试剂盒,购自北京天根生化科技有限公司。凝血指标检测试剂盒由法国Stago公司提供配套试剂。纯化的sheep anti-human FXI IgG由加拿大Cedarlane Laboratories Ltd.公司提供。STA-R全自动血凝仪(法国Stago公司)、PCR扩增仪(ABI Thermalcycler2720)、凝胶电泳仪(美国BIO-rad公司)、凝胶成像系统(上海天能公司)、核酸蛋白分析仪DU800(美国Beckman-Coulter公司)。
1.2.2 引物:根据FXI基因序列(Genbank Number∶NG_008051.1)用Primer Premier 5.0软件设计13对引物(交由上海桑尼生物科技有限公司合成),以覆盖FXI基因所有外显子及其侧翼序列和启动子序列。
1.2.3 样本采集及处理:采集该家系3代8名成员的外周静脉血,以0.109 mol/L枸橼酸钠1∶9抗凝,3 000 r/min离心10 min,上层乏血小板血浆用于检测凝血指标,并于2 h内完成;下层血细胞用于提取基因组DNA。
1.2.4 血浆凝血指标检测:血浆凝血酶原时间(theprothrombin time,PT)、APTT、纤维蛋白原(fibrinogen,FIB)、FXI∶C、凝血因子V III促凝活性(FV III∶C)、凝血因子IX促凝活性(FIX∶C)、凝血因子X II促凝活性(FX II∶C)等凝血指标用一期凝固法在Stago STA-R全自动血凝仪上测定;FXI∶Ag采用酶联免疫吸附测定法检测,所有操作步骤均严格按照试剂说明书进行。
1.2.5 外周血基因组DNA提取:应用酚-氯仿法抽提先证者及家系成员外周血基因组DNA,应用核酸蛋白分析仪DU800检测所提取基因组DNA的浓度和纯度。
1.2.6 PCR扩增及电泳:PCR扩增FXI基因所有外显子及侧翼序列和启动子序列。PCR体系如下:10× PCR buffer 5.0 μL,模板3.0 μL,dNTPs 2.0 μL,引物2.0 μL,Taq DNA聚合酶0.5 μL,ddH2O补足至50 μL。反应条件如下:95 ℃预变性5 min;94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,共30个循环;72 ℃再延伸10 min。
1.2.7 DNA测序分析:PCR产物经1%琼脂糖凝胶电泳进行鉴定,并经纯化后,送上海桑尼生物科技有限公司测序(所用测序仪为ABI PRISM 3730)。通过Chromas软件将测序结果与Genbank数据库中FXI基因序列(NG_008051.1)比对,发现突变位点后,反向测序予以证实。其他家系成员在明确先证者基因突变位点后,PCR扩增相应区域并测序分析。
1.2.8 建立模型分析:基于已知的FXI结构模型,运用Swiss软件对所发现的突变进行分析。
2 结果
2.1 表型检测结果 先证者和其弟弟APTT、FXI∶C、FXI∶Ag均明显异常,分别为78.4 s、2.0%、6.8%和62.1 s、4.5%、10.0%;其家系成员的FXI∶C和 FXI∶Ag均发现有不同程度下降;先证者及家系成员FV III∶C、FIX∶C、FX II∶C等其他凝血指标均无明显异常,见表1。
2.2 FXI基因突变分析 先证者和其弟弟FXI基因第6号外显子的g.15410G>A杂合无义突变导致Trp228stop及第12号外显子的g.25471C>G杂合错义突变导致Cys482Trp;其父亲、姐姐、女儿、外甥女均为Trp228stop的杂合子,而母亲、侄子则为Cys482Trp的杂合子。见表1和图2。
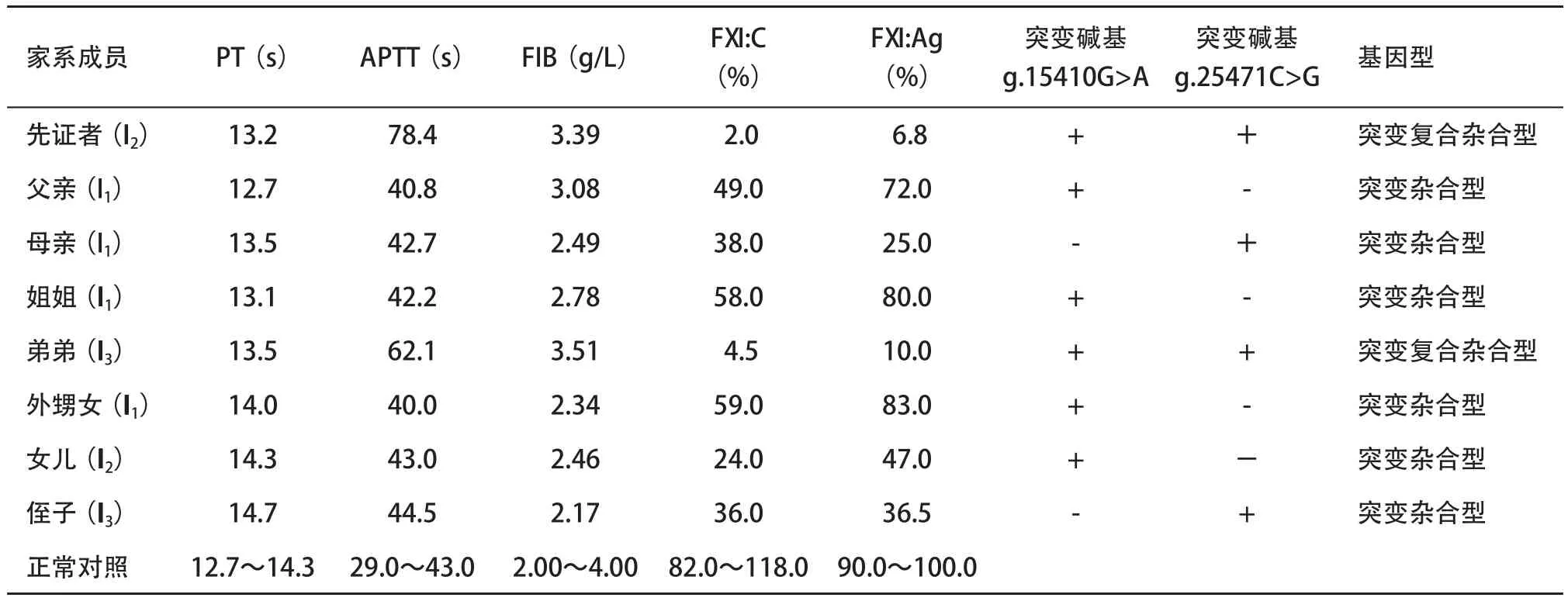
表1 先证者及家系成员实验室检测及基因型结果
2.3 模型建立分析 在野生型蛋白中,Cys482的主干只与Leu571的主干形成一个氢键(如图3绿色虚线所示);当Cys482突变为非极性的Trp时,其主干依然和Leu571的主干形成氢键,但其与His267之间的空间位阻变大(如图3粉色虚线所示)。
3 讨论
遗传性FXI缺陷症是由合成FXI蛋白的基因发生突变引起。该基因位于人类4号染色体上,基因全长23 kb,共含有15个外显子和14个内含子。其中,l号外显子编码5’端非翻译区,2号外显子编码由18个氨基酸组成的信号肽,其余外显子编码成熟肽[4]。合成的FXI蛋白分别由一条轻链和一条重链构成,其中轻链含有238个氨基酸,具有酶催化活性;重链则由4个具有显著特征的苹果状结构域(apple domain,AP)组成。自1989年ASAKAI等[5]首次在犹太人中报道了3种导致遗传性FXI缺陷的基因突变开始,截至2015年,人类基因组数据库(HGMD,http∶//www. hgmd.cf.ac.uk/ac/all.php)共收录了约236种FXI基因突变类型,其中大多数为错义突变或无义突变,而且突变位点覆盖FXI基因所有编码区和调控区。
本研究中,先证者因正常妊娠入住我院,生产过程中出血量较多,并通过输注血浆止血。凝血常规检查显示先证者APTT明显延长,进一步的凝血因子检测发现先证者FXI∶C及FXI∶Ag同步降低,从而诊断为FXI缺陷症。基因分析提示先证者FXI基因6号外显子和12号外显子分别存在无义突变Trp228stop及错义突变Cys482Trp,前者遗传自父亲,后者遗传自母亲。武文漫等[6]首次对无义突变Trp228stop进行了报道,患者也在术前检测时发现FXI缺陷。FXI空间结构显示Trp228残基位于AP3结构域,无义突变产生的截断蛋白缺失AP4及催化活性区,从而蛋白结构完整性受到影响。虽然她的父亲、姐姐、侄女及女儿均携带该无义突变位点,但女儿的FXI∶C及FXI∶Ag相对于其他三者要低更多,考虑到先证者的女儿为新生儿,一般健康新生儿在早期肝功能并不完善,其FXI∶Ag可在6个月后达到正常人水平,因此其抗原及活性水平的降低可能与肝脏合成减少有关[7]。残基Cys482位于蛋白羧基端的丝氨酸蛋白酶区域,其可与残基Cys362形成二硫键连接蛋白的轻链与重链。进一步的模型分析提示,虽然该突变不会影响原本存在的氢键连接,但是当Trp取代Cys后,其会与His267残基之间形成空间位阻。除此之外,错义突变还会影响Cys482残基与Cys362之间二硫键的连接。这些重要结构域的缺失及作用力的改变导致蛋白结构改变,从而形成不稳定蛋白,这可能是导致FXI∶C及FXI∶Ag下降的致病机制,但是具体原因还有待进一步分析。
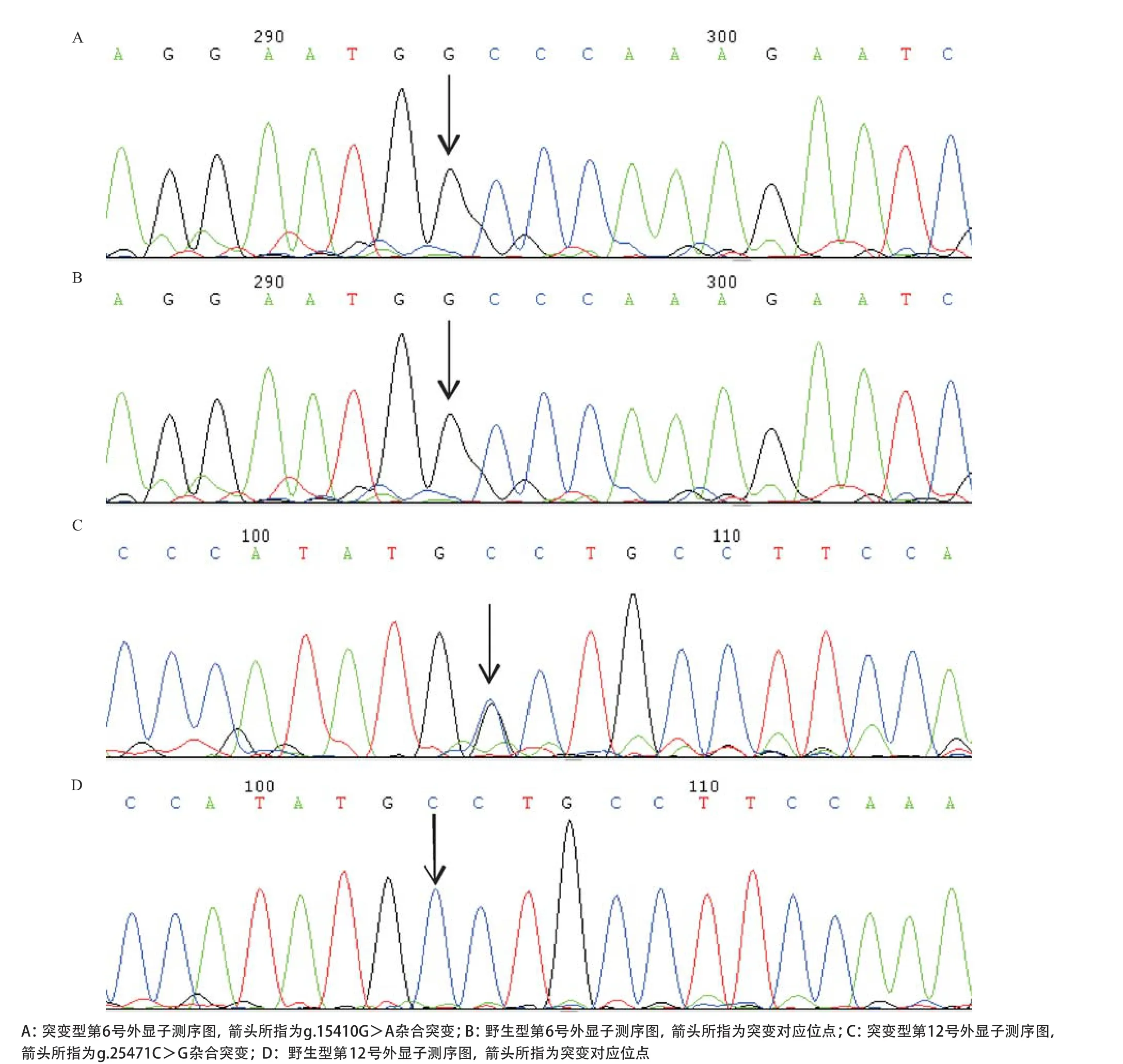
图2 FXI基因测序结果
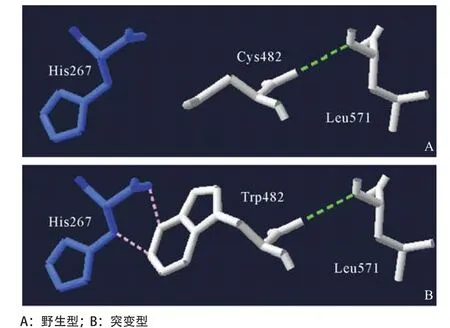
图3 Cys482Trp突变模型
遗传性FXI缺陷症一般无自发性出血表现,多数患者只在严重创伤及术后检查时因明显延长的APTT而被发现。而且患者出血严重程度与APTT延长及FXI缺少程度之间无明显相关性[8],这可能与血小板表面存在的FXI样活性物质,部分代偿了血浆中缺乏的FXI功能有关。目前该病临床治疗上主要以输注血浆纠正凝血功能为主,尚无根治方法。近来研究还提示,FXI是抗血栓治疗的新靶标[9],因此对于遗传性FXI缺陷症的检测、治疗、利用还有待于我们进一步的研究。
[1] BUTENAS S, DEE J D, MANN K G. The function of factor XI intissue factor-initiated thrombin generation[J]. J Thromb Haemost, 2003, 1(10): 2103-2111.
[2] SOUABNI L, MEDDEB N, AJLANI H, et al. Hemarthrosis revealing congenital factor XI de fi ciency[J]. Joint Bone Spine, 2008, 75(3): 348-349.
[3] 武文漫, 王鸿利. 遗传性凝血因子XI缺陷的分子发病机制研究进展[J]. 血栓与止血学, 2004, 10(4): 181-184.
[4] EMSLEY J, MCEWAN P A, GAILANI D. Structure and function of factor XI[J]. Blood, 2010, 115(13): 2569-2577.
[5] ASAKAI R, CHUNG D W, RATNOFF O D, et al. Faetor XI (plasma thromboplastin antecedent) de fi ciency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations[J]. Proc Natl Acad Sci U S A, 1989, 86 (20): 7667-7671.
[6] 武文漫, 王鸿利, 王学锋, 等. 两种新的无义突变Trp228stop和Trp283stop导致的遗传性凝血因子XI缺陷症[J]. 中华血液学杂志, 2003, 24(3): 126-128.
[7] GOMEZ K, BOLTON-MAGGS P. Factor XI de fi ciency[J]. Haemophilia, 2008, 14(6): 1183-1189.
[8] 戴利亚, 张德亭, 谢海啸. 一个遗传性凝血因子XI缺陷症家系的基因分析[J]. 温州医科大学学报, 2015, 45(5): 376-380.
[9] ZHANG H, LÖWENBERG E C, CROSBY J R, et al. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk[J]. Blood, 2010, 116(22): 4684-4692.
(本文编辑:赵翠翠)
Phenotypic diagnosis and genetic analysis in a proband with hereditary coagulation factor XI de fi ciency
YE Jiajia1, YANG Lihong1, HAOXiuping1, CHEN Bicheng2.
1.Department of Laboratory, the First Af fi liated Hospital of Wenzhou Medical University, Wenzhou, 325015; 2.Surgical Laboratory, the First Af fi liated Hospital of Wenzhou Medicatl University, Wenzhou, 325015
Objective: To identify potential mutations and explore the molecular mechanism of a proband underlying hereditary coagulation factor XI (FXI) de fi ciency. Methods: Clinical diagnosis was validated by measuring the prothrombin time (PT), activated partial thromboplastin time (APTT), fi brinogen, FXI activity (FXI:C) and FXI antigen (FXI:Ag). Potential mutations of FXI gene were analyzed by PCR and direct DNA sequencing. Suspected mutations were con fi rmed by sequencing the opposite strand. With the aid of bioinformatics software PyMOL Viewer1.5.x, the wild-type and mutant FXI protein model was also comparatively analyzed. Results: The results of APTT, FXI:C, FXI:Ag in the proband and her brother were obviously abnormal, 78.4 s, 2.0%, 6.8% and 62.1 s, 4.5%, 10.0% respectively. The FXI:C and FXI:Ag of her other family member also presented with different degrees of reduction. A heterozygous g.15410G>A in exon 6 and g.25471C>G in exon 12 were identi fi ed, resulting in the nonsense mutation Trp228stop and missense mutation Cys482Trp. Her father, sister, niece were heterozygous for Trp228stop, while her mother and nephew were for the Cys482Trp mutation. Modeling showed that the missense mutation did not destroy the natural hydrogen bonds, however, it lead to the formation of the space steric hindrance with amino acids 267 which will change the protein structure. Conclusion: A heterozygous Trp228stop mutation and a heterozygous Cys482Trp mutation have been identi fi ed in the FXI gene, which can explain the low levels of the FXI:C and FXI:Ag in the proband.
hereditary coagulation factor XI deficiency; polymerase chain reaction; gene mutation; model analysis
R3554.9
A
10.3969/j.issn.2095-9400.2017.05.009
2016-06-14
温州市科技计划项目(Y20150098)。
叶佳佳(1985-),女,浙江台州人,主管技师,在职硕士生。
陈必成,主任技师,博士生导师,Email:chenbicheng@hotmail.com。
