S6K1 C端自抑制假底物结构域磷酸化与Akt去磷酸化协同调节SP600125诱导巨核细胞系多倍体化
2017-06-09张路遥王丽丽杨金刚邢思宁赵松于颖谢晓冬马东初
张路遥,王丽丽,杨金刚,邢思宁,赵松,于颖,谢晓冬,马东初
1.辽宁中医药大学,辽宁 沈阳 113021;2.沈阳军区总医院 全军肿瘤诊治中心医学实验科,辽宁 沈阳 110016;3.沈阳军区总医院 全军肿瘤诊治中心肿瘤科,辽宁 沈阳 110016
S6K1 C端自抑制假底物结构域磷酸化与Akt去磷酸化协同调节SP600125诱导巨核细胞系多倍体化
张路遥1,3,王丽丽2,杨金刚2,邢思宁2,赵松2,于颖2,谢晓冬3,马东初2
1.辽宁中医药大学,辽宁 沈阳 113021;2.沈阳军区总医院 全军肿瘤诊治中心医学实验科,辽宁 沈阳 110016;3.沈阳军区总医院 全军肿瘤诊治中心肿瘤科,辽宁 沈阳 110016
目的:探讨JNK抑制剂SP600125诱导巨核细胞白血病系多倍体化的调控机制。方法:用SP600125和3种抑制剂(PD184352、U0126和LY294002)处理Dami和CMK细胞,用点突变技术对核糖体蛋白S6激酶1(S6K1)C端自抑制假底物结构域和疏水基序突变构建的S6K1质粒转染细胞,流式细胞术分析细胞的DNA倍性,Western印迹检测S6K1、MAPK和Akt蛋白的表达及磷酸化修饰位点的变化。结果:当单独使用3种抑制剂时,对Dami和CMK细胞的倍性无影响。当SP600125与3种抑制剂联合时,尽管PD184352下调了p44/42 MAPK在Thr202/Tyr204位点的磷酸化,但其并不抑制SP600125诱导的Dami和CMK细胞的多倍体化;相反,U0126抑制SP600125诱导的Dami和CMK细胞的多倍体化,但并不下调p44/42 MAPK的磷酸化;而LY294002增加Akt的磷酸化,并阻断SP600125诱导的Dami和CMK细胞的多倍体化。这3种抑制剂均在SP600125诱导的多倍体Dami和CMK细胞中部分抑制S6K1的Thr421/Ser424磷酸化,但并不增加S6K1的Thr389磷酸化。转染S6K1突变质粒的Dami细胞,并没有对SP600125诱导的多倍体化产生影响,同时,无论是模拟磷酸化或去磷酸化的Thr389突变均对SP600125诱导的多倍体化无影响,且未显现出与LY294002的协同作用。C端自抑制假底物结构域突变质粒(S6K1-D3E)可以进一步阻断SP600125诱导的已经被LY294002部分阻断的Dami细胞多倍体化。结论:S6K1的C端自抑制假底物结构域磷酸化与Akt去磷酸化协同调节SP600125诱导巨核细胞白血病系的多倍体化。
核糖体蛋白S6激酶1(S6K1);Akt;巨核细胞;多倍体化
巨核细胞是产生血小板的前体细胞,多倍体化在巨核细胞发育过程中发挥重要作用[1-3]。我们前期研究发现,核糖体蛋白S6激酶1(ribosomal protein S6 kinase 1,S6K1)参与Nocodazole(微管抑制剂)和SP600125(JNK抑制剂)诱导的巨核细胞白血病细胞系Dami和CMK多倍体化,伴随着S6K1在Thr421/Ser424位点的磷酸化及Thr389位点的去磷酸化[4-5],并且H-89阻断SP600125诱导的Dami和CMK细胞多倍体化伴随着S6K1在Thr389位点磷酸化的增加,以及在Thr421/Ser424位点磷酸化的减少[5]。然而,S6K1在药物诱导巨核细胞白血病细胞系多倍体化过程中的作用机制尚不十分清楚。另有报道脯氨酸介导有丝分裂原调节丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPK)磷酸化Thr421/Ser424,其位于S6K1的C端自抑制假底物结构域[6]。此外,Akt,又称蛋白激酶B(protein kinase B,PKB),通过哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex-1,mTORC1)激活S6K1[7]。
本此,我们用PD184352(高效选择性MEK1/2抑制剂)、U0126(MEK抑制剂)和LY294002(PI3K抑制剂)处理Dami和CMK细胞,用点突变技术构建S6K1 C端自抑制假底物结构域和疏水基序内不同位点的突变质粒,探讨了S6K1与MAPK和Akt通路在SP600125诱导细胞多倍体化方面所起的作用。我们的研究表明,S6K1的C端自抑制假底物结构域磷酸化与Akt去磷酸化协同调节SP600125诱导巨核细胞白血病系的多倍体化。
1 材料与方法
1.1 材料
人巨核细胞白血病细胞系Dami购自美国国立细胞库(ATCC,其编号为CRL-9792),CMK细胞从德国微生物和细胞培养保存中心获得。二甲基亚砜(DMSO)、碘化丙啶(PI)购自Sigma公司;SP600125购自美国LC实验室;PD184352、U0126、LY294002购自 Selleckchem公司;RPMI 1640培养基购自Life Technologies公司;新生牛血清购自杭州四季青生物工程材料有限公司;兔抗人S6K1抗体、兔抗人磷酸化S6K1(Thr389)抗体、兔抗人磷酸化S6K1(Thr421/Ser424)抗体、兔抗人p44/42 MAPK抗体、兔抗人磷酸化p44/42 MAPK(Thr202/Tyr204)抗体、兔抗人Akt抗体,兔抗人磷酸化Akt(Ser473)抗体、辣根过氧化物酶(HRP)标记的山羊抗兔IgG和HRP标记的山羊抗鼠IgG均购自 Cell Signaling Technology公司;β-actin购自Santa Cruz Biotechnology公司;Amersham ECL Prime Western印迹试剂盒购自GE Healthcare公司;即用PCR扩增试剂盒和SanPrep柱式质粒DNA小量抽提试剂盒购自生工生物工程(上海)有限公司;限制性核酸内切酶DpnⅠ和大肠杆菌DH5α感受态细胞购自TaKaRa公司;Amaxa核转染仪和转染试剂盒购自Amaxa公司。
1.2 细胞培养与处理
CMK和Dami细胞均以2×105/mL的密度接种于T75培养瓶中,在含10%新生牛血清的培养液中生长,将培养瓶置于37℃、5%CO2的潮湿孵箱中培养。在3种抑制剂诱导倍体化实验中,分别将PD184352(2μmol/L)、U0126(10μmol/L)或LY294002(30μmol/L)加入培养基,连续培养72 h。在SP600125联合3种抑制剂诱导多倍体化实验中,在加入SP600125(32μmol/L)前1 h,分别将PD184352(2μmol/L)、U0126(10μmol/L)或LY294002(30μmol/L)加入培养基,连续培养72 h。以终浓度为1%的DMSO作为溶媒对照组。孵育后,收集细胞,并用PBS洗涤3次。
1.3 细胞倍性分析
分别收集实验组和对照组细胞5×105个,用80%冰甲醇于-20℃固定过夜,PBS洗涤3次,加入50 mg/mL PI染液,室温避光孵育30 min,用流式细胞仪检测分析DNA倍性。
1.4 Western印迹
收集细胞,加入含蛋白酶抑制剂的细胞裂解液,冰浴中超声波破碎细胞,100℃煮沸5 min,4℃、12 000 r/min离心5 min,回收上清即为提取的蛋白样品,采用二辛可宁酸(BCA)法测定蛋白浓度。取蛋白样品上样进行SDS-PAGE,并湿转至硝酸纤维素膜上,将膜在含5%脱脂牛奶的TBS-T中室温孵育2 h。兔抗人S6K1抗体(1∶5000)、兔抗人p-S6K1(Thr389)抗体(1∶300)、兔抗人p-S6K1(Thr421/Ser424)抗体(1∶500)、兔抗人p44/ 42 MAPK抗体(1∶5000)、兔抗人磷酸化p44/42 MAPK(Thr202/Tyr204)抗体(1∶500)、兔抗人Akt抗体(1∶2000)、兔抗人磷酸化 Akt(Ser473)抗体(1∶300)、小鼠抗人β-actin抗体(1∶80 000)孵育,4℃摇床振荡过夜;TBS-T洗涤3次,加入HRP标记的山羊抗兔IgG(1∶40 000)或HRP标记的山羊抗小鼠IgG(1∶40 000),室温孵育2 h;TBS-T洗涤3次,暗室内用ECL化学发光检测,X线胶片曝光。
1.5 定点突变
设计含S6K1相应位点碱基突变的一对互补引物,采用PAGE纯化方式由Invitrion公司合成,按照要求加入一定量的无菌水溶解引物,配置成100μmol/L的母液,使用时稀释至1/10。采用即用PCR扩增试剂盒,反应体系包括模板质粒1 μL(50 ng)、2条引物各1μL、水9.5μL、2×HiFi缓冲液12.5μL。PCR条件为94℃预变性1 min,94℃变性40 s、60℃退火1 min、68℃延伸7 min,进行18个循环。PCR后加入1μLDpnⅠ酶,37℃酶切1 h,酶切产物10μL直接转化DH5α感受态细胞,在氨苄西林平皿中筛选,挑取单克隆进行质粒抽提和测序,以确认突变位点碱基取代是否正确,并且没有其他突变被导入。T389E突变用引物1进行;T389A突变用引物2进行;D3E突变用2对引物,先用引物3进行S418D+T421E+S424D突变,确定突变成功后,以突变质粒为模板,用引物4进行S411D突变;T389E/D3E突变用引物1,以S6K1-D3E为模板进行;T389A/D3E突变用引物2,以S6K1-D3E为模板进行。所有突变体见表1。
1.6 核转染
Dami细胞用编码S6K1-WT或S6K1-T389E、S6K1-T389A、S6K1-D3E、S6K1-T389E/D3E和S6K1-T389A/D3E突变体的质粒瞬时转染,参见前文[4]。简言之,用预冷的PBS缓冲液洗涤Dami细胞,然后将细胞重悬于指定的电穿孔缓冲液中,细胞悬液的终浓度为1.2×108/mL。每种突变质粒2μg用0.1 mL细胞悬液混匀,将混合物转移至一个2.0 mm电穿孔池中,用Amaxa核转染仪进行核转染。转染后,将细胞重悬于含10%胎牛血清的RPMI1640培养基中,孵育过夜。孵育后,转染的Dami细胞用SP600125(32μmol/L)在有或无LY294002(30μmol/L)预处理1 h后,于37℃、5%CO2的潮湿孵箱中诱导72 h。孵育72 h后收集细胞,检测细胞倍性。以未转染突变质粒,但用SP600125诱导的Dami细胞作为对照组。
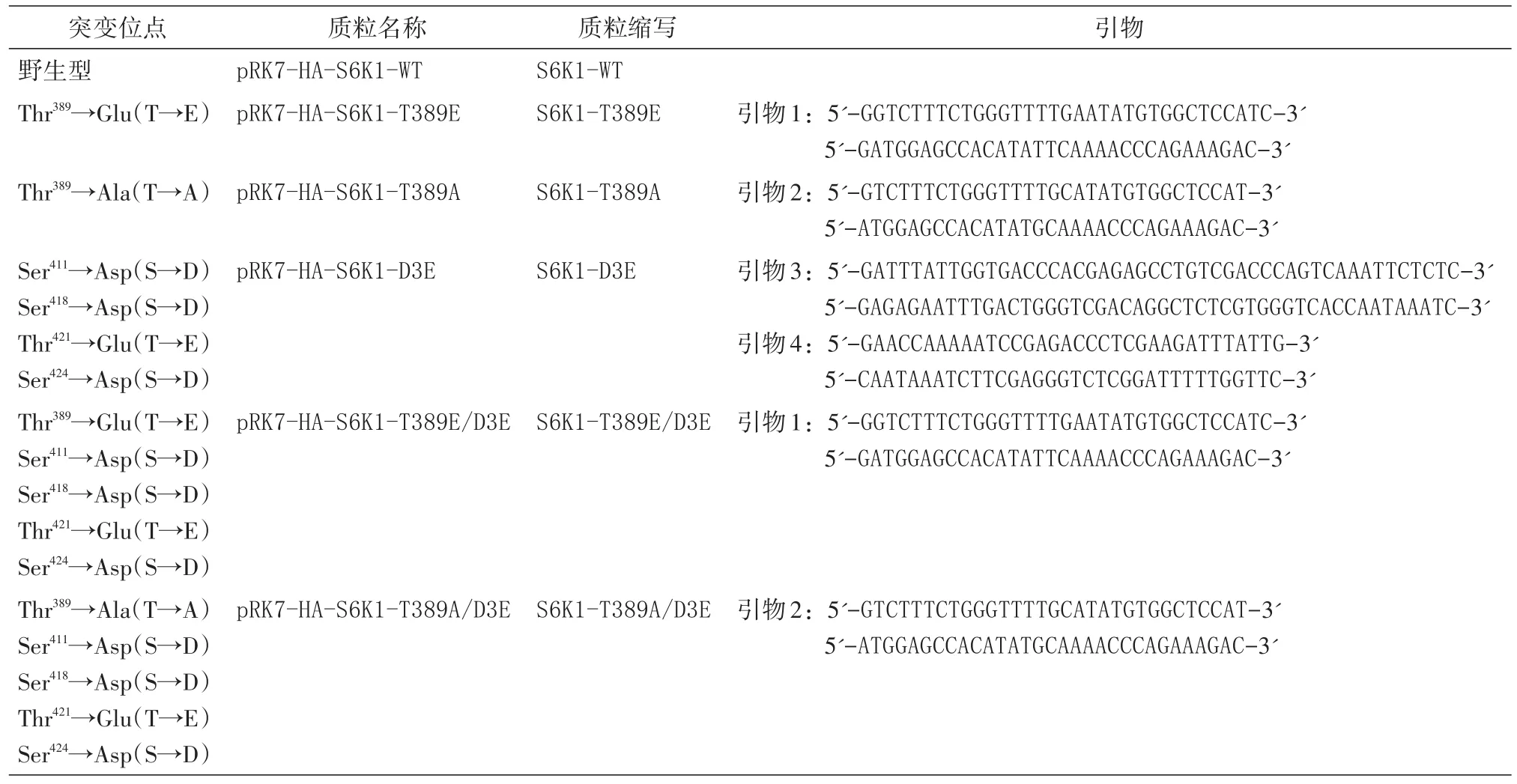
表1 通过点突变产生的S6K1突变质粒
1.7 统计学分析
所有实验均重复3次。采用SPSS13.0统计软件分析实验数据,数据用x±s表示,进行t检验,P<0.05为差异具有统计学意义。
2 结果
2.1 PD184352、U0126和LY294002对巨核细胞系多倍体化的影响
用PD184352(高效选择性MEK1/2抑制剂)、U0126(MEK抑制剂)和LY294002(PI3K抑制剂)来研究这些通路是否参与SP600125诱导Dami和CMK细胞多倍体。基于 PD184352、U0126和LY294002对底物的选择抑制浓度(数据未示),对应的使用终浓度分别为2、10和30μmol/L。
与我们以往报道的结果一致,SP600125处理显著增加Dami和CMK细胞的DNA多倍性。与对照组(DMSO处理)Dami和CMK细胞的DNA平均倍性(2.8±0.2和2.9±0.4)相比,SP600125处理组的Dami和CMK细胞DNA平均倍性分别增至9.3±0.6和9.9±0.2(图1A、B)。意外的是PD184352不能抑制SP600125诱导Dami和CMK细胞多倍体化。PD184352与SP600125联合处理Dami和CMK细胞的DNA平均倍性与SP00125单独处理没有明显差别,分别为8.9±0.7和9.5±0.3(图1A、B)。然而,U0126和LY294002分别部分阻断SP6000125诱导Dami和 CMK细胞 的多倍体化。U0126与SP600125联合处理导致Dami和CMK细胞的DNA平均倍性分别降至6.8±0.2和7.3±0.3(与SP00125处理组比,P<0.05)。LY294002与SP600125联合处理导致Dami和CMK细胞DNA平均倍性分别降至6.6±0.1和7.2±0.2(与SP00125处理组比,P<0.05)。同时,这些抑制剂自身并不能对Dami和CMK细胞的DNA含量分布产生影响(图1C,CMK细胞数据未示)。该结果提示,PI3K和MAPK信号途径,而不是MEK1/2信号途径,可能参与SP600125诱导Dami和CMK细胞的多倍体化。
2.2 LY294002通 过 上 调 Akt磷 酸 化 阻 断SP600125诱导的多倍体化

图1 流式细胞仪分析SP600125单独处理或SP600125分别联合PD184352、U0126和LY294002处理Dami和CMK细胞72 h,以及3种抑制剂PD184352、U0126和LY294002分别处理Dami细胞72 h的倍性变化
为进一步探讨PD184352、U0126和LY294002对相关信号转导通路的影响,用Western印迹分析了相关信号分子的表达和修饰变化。如图2所示,PD184352可以显著抑制SP600125诱导的p44/ 42 MAPK的Thr202/Tyr204的磷酸化,U0126则无此作用,表明SP600125诱导的Dami和CMK细胞多倍体化不依赖于MAPK信号转导路径。LY294002可上调SP600125诱导多倍体化Dami和CMK细胞Akt Ser473的磷酸化,表明 PI3K/Akt通路在用LY294002预处理的SP600126诱导的多倍体Dami和CMK细胞中被激活。鉴于PI3K/Akt通路活化促进细胞增殖,且LY294002处理可部分阻断SP600125诱导的Dami和CMK细胞多倍体化,以及SP600125轻度下调Akt Ser473磷酸化(尽管并不十分明显),因此,确保PI3K/Akt通路不过度活化有利于SP600125诱导Dami和CMK细胞多倍体化。值得注意的是,PD184352、LY294002和U0126均在SP600125诱导的多倍体Dami和CMK细胞中,部分抑制S6K1的Thr421/Ser424磷酸化。然而,这些抑制剂并不增加S6K1的Thr389磷酸化。这些数据表明S6K1单独的Thr421/Ser424磷酸化可能不足以介导由SP600125诱导Dami和CMK细胞的多倍体化,其他信号通路可能参与此过程。
2.3 S6K1 C端自抑制假底物结构域磷酸化与Akt去磷酸化协同调节SP600125诱导的多倍体化

图2 Western印迹检测SP600125单独处理和SP600125分别联合PD184352、U0126、LY294002处理Dami和CMK细胞72 h的蛋白表达及磷酸化修饰位点的变化
我们以往曾发现myc-d/ED3E-pRK5(包含Ser411→Asp、Ser418→Asp、Thr421→Glu、Ser424→Asp和Thr389→Glu突变质粒)可以阻断Nocodazole诱导的Dami细胞多倍体化[4]。SP600125诱导 Dami和CMK细胞多倍体化伴随着S6K1的Thr421/Ser424磷酸化和Thr389去磷酸化(图2),而且,H-89通过占据S6K1的ATP结合位点,下调Thr421/Ser424磷酸化和上调Thr389磷酸化,阻断SP600125诱导Dami和CMK细胞多倍体化[5]。然而,PD184352虽然可以下调SP600125诱导多倍体化Dami和CMK细胞上Thr421/Ser424磷酸化,但其并不阻断SP600125诱导Dami和CMK细胞的多倍体化(图1、2)。因此,为了明确S6K1 C端自抑制假底物结构域磷酸化和Thr389磷酸化修饰在SP600125诱导多倍体化方面所起的作用,我们构建了C端自抑制假底物结构域内相关磷酸化位点和Thr389位点的突变质粒,即S6K1-WT、S6K1-T389E、S6K1-T389A和S6K1-D3E(图3),并分别用这些质粒转染Dami细胞,观察了对SP600125诱导Dami细胞倍体化的影响。令我们意外的是,与SP600125诱导的Dami细胞倍体化(7.04±0.37)相比,无论是转染野生型S6K1(S6K1-WT:7.27±0.37)或突变型 S6K1(S6K1-T389E:6.93±0.32;S6K1-T389A:6.86±0.51;S6K1-D3E:6.78±0.4)均不能影响SP600125诱导的Dami细胞倍体化。然而,当S6K1-D3E与LY294002共同处理SP600125诱导的Dami细胞后,LY294002对倍体化的抑制作用(从7.04±0.37降至5.30± 0.18,P<0.05)可被 S6K1-D3E进一步增强(从5.30±0.18降至4.07±0.14,P<0.05)(图4A、B)。然而,无论是S6K1-D3E突变联合Thr389位点的模拟磷酸化突变(S6K1-T389E/D3E),还是去磷酸化突变(S6K1-T389A/D3E)均未明显增强或降低S6K1-D3E对SP600125诱导倍体化的抑制作用(图4C)。鉴于LY294002诱导Akt的Ser473磷酸化,该结果提示S6K1 C端自抑制假底物结构域的磷酸化参与SP600125诱导Dami细胞的多倍体化调控,并需要Akt Ser473去磷酸化的协同作用。
3 讨论
哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路是调控细胞合成代谢的重要开关[8],已有研究发现,其参与哺乳动物细胞的多倍体化调控[9-10]。S6K1是mTOR下游的重要靶分子之一,其通过调控核糖体合成、蛋白质合成、细胞周期进程和代谢,在细胞生长、增殖和分化中起重要作用[11-15]。我们以往的研究发现,S6K1可能在药物诱导巨核细胞白血病细胞系多倍体化过程中发挥重要作用[4-5]。然而,详细的作用机制尚不十分清楚,而且S6K1活性的调控比较复杂,至少包括mTOR、生长因子、激素和应激有关的信号转导途径调控的8个磷酸化位点(即 Thr229、Ser371、Thr389、Ser404、Ser411、Ser418、Thr421和Ser424)[16-18]。因此,在前期研究基础上,我们进一步解析了S6K1的磷酸化修饰变化在药物诱导巨核细胞白血病细胞系多倍体化过程中的调控机制。有研究报道,血清素诱导肺动脉平滑肌生长,且15(S)-羟基花生四烯酸诱导血管生成需要S6K1在Thr421/Ser424位点的磷酸化,但该过程可以被LY294002显著阻断[19-20];脯氨酸介导有丝分裂原调节MAPK磷酸化Thr421/Ser424[6];在心肌细胞中U0126阻断TPA或胰岛素诱导的S6K1 Thr421/Ser424位点的磷酸化[21]。
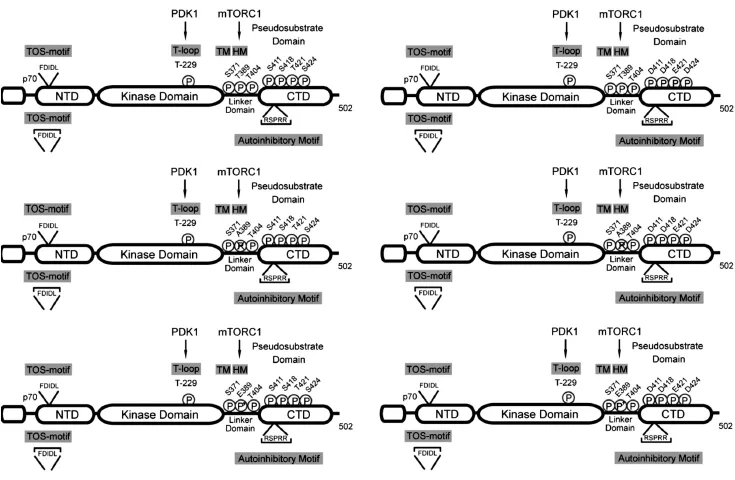
图3 S6K1突变质粒结构图
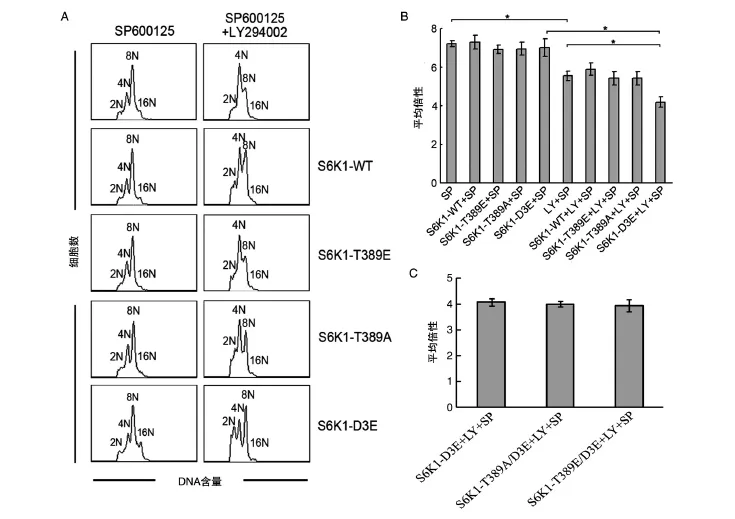
图4 S6K1突变质粒对SP600125单独处理和SP600125联合LY294002处理Dami细胞72 h的倍性影响
在本研究中,我们发现SP600125诱导p44/42 MAPK在Thr202/Tyr204位点的磷酸化及Dami和CMK细胞的多倍体化。虽然PD184352和U0126均能显著抑制S6K1在Thr421/Ser424的磷酸化,然而尽管PD184352下调了p44/42 MAPK在Thr202/Tyr204位点的磷酸化,但并不抑制SP600125诱导的Dami和CMK细胞的多倍体化。与此相反,U0126抑制SP600125诱导的Dami和CMK细胞的多倍体化,但并不下调p44/42 MAPK的磷酸化。这些结果表明SP600125诱导的Dami和CMK细胞多倍体化不依赖于MAPK信号转导路径。有趣的是,LY294002不仅抑制Thr421/Ser424的磷酸化,同时增加Akt的磷酸化,并阻断SP600125诱导的Dami和CMK细胞的多倍体化,该结果提示LY294002可能通过上调Akt磷酸化阻断SP600125诱导的多倍体化。值得注意的是,这3种抑制剂均在SP600125诱导的多倍体Dami和CMK细胞中部分抑制S6K1的Thr421/Ser424磷酸化,然而,它们并不增加S6K1的Thr389磷酸化。总之,上述结果提示S6K1在Thr421/Ser424位点的磷酸化可能在SP600125诱导Dami和CMK细胞的多倍体化中发挥重要作用,然而,S6K1单独的Thr421/Ser424磷酸化可能不足以介导由SP600125诱导Dami和CMK细胞的多倍体化。
有文献报道,通过复合多位点磷酸化S6K1的逐步活化是通过C端假底物结构域中的4个位点(Ser411、Ser418、Thr421和Ser424)的磷酸化来启动,其诱导构象变化、疏水基序(HM)和T-loop位点[7,22]。虽然这4个C端位点的磷酸化有助于S6K1激活,但其并非关键。这4个位点突变为丙氨酸残基或者从C端删除101个氨基酸残基可以适当地诱导S6K1激活[7]。一般认为,Thr389位点的磷酸化是S6K1活化的标志[23]。尽管前期研究表明S6K1参与巨核细胞多倍体化的调控[4-5],但调控S6K1活性的磷酸化位点的磷酸化修饰和倍体化的关系尚不清楚。在本研究中,我们发现用S6K1-WT、S6K1-T389E、S6K1-T389A和S6K1-D3E分别转染Dami细胞,并没有对SP600125诱导的多倍体化Dami细胞产生影响,同时,具有Thr389→Glu突变或Thr389→Ala突变的任何一种S6K1对SP600125单独处理或联合LY294002处理的Dami细胞多倍体化没有影响。然而,S6K1-D3E可以进一步阻断SP600125诱导的已被LY294002部分阻断的Dami细胞多倍体化。鉴于LY294002诱导Akt的Ser473磷酸化,阻断SP00125诱导的多倍体化,而且S6K1-D3E包含Ser411→Asp、Ser418→Asp、Thr421→Glu和Ser424→Asp的突变位点均位于C端自抑制假底物结构域内,提示Akt的Ser473位点去磷酸化和C端自抑制假底物结构域内上述位点的磷酸化在SP600125诱导的多倍体化方面发挥重要作用。然而,S6K1蛋白可分成数个重要结构域,其通过复杂的多位点磷酸化来调控其活性。因此,仍需要进一步的研究来解析巨核细胞白血病细胞系多倍体化期间S6K1的相关分子机制。
[1]Wilcox D A.Megakaryocyte-and megakaryocyte precursor-related gene therapies[J].Blood,2016,127(10): 1260-1268.
[2]Geddis A E. Megakaryopoiesis[J]. Semin Hematol, 2010,47(3):212-219.
[3]Trakala M,Rodríguez-Acebes S,Maroto M,et al. Functional reprogramming of polyploidization in megakaryocytes[J].Dev Cell,2015,32(2):155-167.
[4]Ma D,Yu H,Lin D,et al.S6K1 is involved in polyploidization through its phosphorylation at Thr421/ Ser424[J].J Cell Physiol,2009,219(1):31-44.
[5]王丽丽,杨金刚,李长岭,等.核糖体蛋白S6激酶1(S6K1)介导SP600125诱导的巨核细胞白血病细胞系多倍体化依赖于细胞分化程度[J].细胞与分子生物学杂志,2016,32(10):1336-1341.
[6]Mukhopadhyay N K,Price D J,Kyriakis J M,et al. An array of insulin-activated,proline-directed serine/ threonine protein kinases phosphorylate the p70 S6 kinase[J].J Biol Chem,1992,267:3325-3335.
[7]Magnuson B,Ekim B,Fingar D C.Regulation and function of ribosomal protein S6 kinase(S6K)within mTOR signalling networks[J].Biochem J,2012,441:1-21.
[8]Li T,Wang G.Computer-aided targeting of the PI3K/ Akt/mTOR pathway:toxicity reduction and therapeutic opportunities[J].Int J Mol Sci,2014,15(10):18856-18891.
[9]Li Y,Chen X,Tang X,et al.DNA synthesis during endomitosis is stimulated by insulin via the PI3K/Akt and TOR signaling pathways in the silk gland cells of Bombyx mori[J].Int J Mol Sci,2015,16(3):6266-6280.
[10]Sharma S,Yao H P,Zhou Y Q,et al.Prevention of BMS-777607-induced polyploidy/senescence by mTOR inhibitor AZD8055 sensitizes breast cancer cells to cytotoxic chemotherapeutics[J].Mol Oncol,2014,8(3):469-482.
[11]Tavares M R,Pavan I C,Amaral C L,et al.The S6K protein family in health and disease[J].Life, 2015,131:1-10.
[12]Yi S A,Um S H,Lee J,et al.S6K1 Phosphorylation of H2B mediates EZH2 trimethylation of H3:A determinant of early adipogenesis[J].Mol Cell,2016,62(3): 443-452.
[13]Wu H,Xiao Z,Wang K,et al.MiR-145 is downregulated in human ovarian cancer and modulates cell growth and invasion by targeting p70S6K1 and MUC1 [J].Biochem Biophys Res Commun,2013,441(4):693-700.
[14]Pavan I C,Yokoo S,Granato D C,et al.Different interactomes for p70-S6K1 and p54-S6K2 revealed by proteomic analysis[J]. Proteomics, 2016,16(20):2650-2666.
[15]Miyake S,Wakita H,Bernstock J D,et al.Hypophosphorylation of ribosomal protein S6 is a molecular mechanism underlying ischemic tolerance induced by either hibernation or preconditioning[J].J Neurochem, 2015,135(5):943-957.
[16]Shin S,Wolgamott L,Yu Y,et al.Glycogen synthase kinase(GSK)-3 promotes p70 ribosomal protein S6 kinase(p70S6K)activity and cell proliferation[J].Proc Natl Acad Sci USA,2011,108(47):E1204-1213.
[17]Wang J,Zhong C,Wang F,et al.Crystal structures of S6K1 provide insights into the regulation mechanism of S6K1 by the hydrophobic motif[J].Biochem J,2013,454(1):39-47.
[18]Zhang H,Hoff H,Marinucci T,et al.Mitogen-independent phosphorylation of S6K1 and decreased ribosomal S6 phosphorylation in senescent human fibroblasts[J].Exp Cell Res,2000,259(1):284-292.
[19]Liu Y,Fanburg B L.Serotonin-induced growth of pulmonary artery smooth muscle requires activation of phosphatidylinositol 3-kinase/serine-threonine protein kinase B/mammalian target of rapamycin/p70 ribosomal S6 kinase 1[J].Am J Respir Cell Mol Biol,2006, 34:182-191.
[20]Ohanna M,Sobering A K,Lapointe T,et al.Atrophy of S6K1(2/2)skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control[J].Nat Cell Biol,2005,7:286-294.
[21]Iijima Y,Laser M,Shiraishi H,et al.c-Raf/MEK/ ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells[J].J Biol Chem,2002,277:23065-23075.
[22]Fenton T R,Gout I T.Functions and regulation of the 70kDa ribosomal S6 kinases[J].Int J Biochem Cell Biol,2011,43(1):47-59.
[23]Doscas M E,Williamson A J,Usha L,et al.Inhibition of p70 S6 kinase(S6K1)activity by A77 1726 and its effect on cell proliferation and cell cycle progress[J].Neoplasia,2014,16(10):824-834.
SP600125-Induced Polyploidization of Megakaryocytic Cell Lines by the Regulation of S6K 1 C-Terminal Autoinhibitory Pseudosubstrate Domain Phosphorylation and Akt Dephosphorylation
ZHANG Lu-Yao1,3,WANG Li-Li2,YANG Jin-Gang2,XING Si-Ning2, ZHAO Song2,YU Ying2,XIE Xiao-Dong3*,MA Dong-Chu2*
1.Liaoning University of Chinese Medicine,Shenyang 113021;
2.Department of Experimental Medicine,Tumor Center of PLA,Norhern Hospital,Shenyang 110016;
3.Department of Oncology,Tumor Center of PLA,Norhern Hospital,Shenyang 110016;China
*Co-corresponding authors,MA Dong-Chu,E-mail:mdc580819@sina.com;XIE Xiao-Dong,E-mail:doctor-xxd@163.com
[Abstract]Objective:To investigate the mechanism of SP600125-induced polyploidization of megakaryocytic leukemia cell lines.Methods:Dami and CMK cells were treated with SP600125 and three inhibitors PD184352, U0126 and LY294002.Meanwhile,we constructed plasmid encoding mutated ribosomal protein S6 kinase 1(S6K1)by point mutagenesis targeting the C-terminal autoinhibitory pseudosubstrate domain and the hydrophobic motif of S6K1 to transfecte cells.The DNA ploidy was analyzed by flow cytometry.The expression and phosphorylation of S6K1,MAPK and Akt were detected by Western blot.Results:When three inhibitors were used alone,there was no effect on the ploidy of Dami and CMK cells.When SP600125 was combined with three inhibitors,PD184352 did not inhibit SP600125-induced polyploidization of Dami and CMK cells,although it decreased the phosphorylation of p44/42 MAPK at Thr202/Tyr204.In contrast,U0126 inhibited SP600125-induced polyploidization of Dami and CMK cells,but did not decreased p44/42 MAPK phosphorylation.While LY294002 increased the phosphorylation of Akt and blocked SP600125-induced polyploidization of Dami and CMK cells.Notably,these three inhibitors,in SP600125-induced polyploidization of Dami and CMK cells,partially inhibited the phosphorylation of S6K1 at Thr421/Ser424,but did not increase the phosphorylation of S6K1 at Thr389.Dami cells that transfected with S6K1 mutant plasmid did not affect SP600125-induced polyploidization.At the same time,either the phosphorylation or dephosphorylation mutations with Thr389sites had no effect on SP600125-induced polyploidization,and it did not show synergistic effect with LY294002.However,C-terminal autoinhibitory pseudosubstrate domain mutant plasmid(S6K1-D3E)could further block polyploidization of SP600125-induced polyploidization of Dami cells that had been partially blocked by LY294002.Conclusion:SP600125-induced polyploidization of megakaryocytic leukemia cell lines by the regulation of S6K1 C-terminal autoinhibitory pseudosubstrate domain phosphorylation and Akt dephosphorylation.
S6 kinase 1(S6K1);Akt;megakaryocytes;polyploidization
Q25
A
1009-0002(2017)03-0233-09
10.3969/j.issn.1009-0002.2017.03.002
2016-12-29
国家自然科学基金(61302003,31571398)
张路遥(1990-),女,硕士研究生,(E-mail)329730208@qq.com
马东初,(E-mail)mdc580819@sina.com;谢晓冬,(E-mail)doctor-xxd@163.com
