咔唑类化合物在MoS2晶体表面吸附行为的第一性原理
2017-06-05杨敬一周秀欢袁佩青徐心茹
杨敬一, 何 萧, 周秀欢, 袁佩青, 徐心茹
(华东理工大学 化工学院, 上海 200237)
咔唑类化合物在MoS2晶体表面吸附行为的第一性原理
杨敬一, 何 萧, 周秀欢, 袁佩青, 徐心茹
(华东理工大学 化工学院, 上海 200237)
基于密度泛函理论,采用VASP方法研究了咔唑类化合物(咔唑、3-甲基咔唑和1,5-二甲基咔唑)在MoS2(0 0 -3)晶体表面的吸附行为,考察引起其吸附能差异的原因。结果表明,咔唑类化合物在MoS2晶体表面的吸附过程中为电子受体,其吸附能随MoS2片层数的增多而减弱;由于空间位阻和电子效应的共同作用,使得咔唑类化合物在MoS2晶体表面的吸附能随着苯环上甲基取代数目的增加而变小;Bader电荷及高低占据轨道的分析表明,咔唑类化合物分子上N原子得电子多少的顺序与它们在MoS2晶体表面的吸附能大小顺序一致,吡咯环N原子上的电子流向催化剂Mo原子,同时咔唑类化合物分子的LUMO轨道接受S原子HOMO轨道的电子形成反馈键,使得咔唑类化合物在MoS2晶体表面稳定吸附。
咔唑类化合物; MoS2晶体; 吸附; 密度泛函理论; Bader电荷
原油日趋重质化劣质化,加氢工艺成为生产清洁车用燃料的主要方法。在深度加氢脱硫过程中,原料油中的有机含氮化合物使加氢催化剂中毒,抑制加氢脱硫反应活性,严重影响产品质量,特别在生产国Ⅴ和国Ⅵ汽柴油时,影响尤为显著[1-2]。
大部分原油和馏分油中的氮以有机杂环含氮化合物的形式存在,包括以喹啉、吡啶为代表的碱性含氮化合物和以咔唑、吲哚为代表的非碱性含氮化合物[3]。国内外众多学者基于密度泛函理论(DFT)研究了含氮化合物在加氢脱硫催化剂表面的吸附作用。Sun等[4-5]利用密度泛函理论计算了吡啶、喹啉、吖啶、吡咯、吲哚和咔唑在NiMoS晶体表面的吸附能,结果表明,碱性氮化物在晶体表面以垂直吸附为主,而非碱性氮化物只能平行吸附在晶体表面,且碱性含氮化合物的吸附能大于非碱性含氮化合物。孙炜等[6]同样针对NiMoS晶体模型,选取广义梯度近似(GGA)中的PW91交换关联泛函计算了吡啶、喹啉、咔唑和吲哚的吸附构型和吸附能,结果表明,吡啶和喹啉是通过Ni-N键垂直吸附在晶体表面,而吲哚通过吡咯环的β-C键吸附在晶体表面,咔唑则主要通过苯环吸附在晶体表面,吲哚和咔唑均以平行吸附为主。Abdallah等[7]采用DFT理论研究了吡咯在Mo(110)表面的平行、垂直和倾斜吸附,结果表明,吡咯以平行吸附构型为主,不可能垂直吸附在Mo表面;多个吡咯吸附在Mo表面时,以倾斜吸附为主。Temel等[8]利用DFT理论结合扫描隧道显微镜(STM),研究了吡啶分子在MoS2团簇表面的吸附作用,结果表明,吡啶分子本身与MoS2团簇的相互作用很弱,但在有H2存在的情况下,吡啶分子在MoS2团簇边缘稳定吸附。Ren等[9]为了深入研究加氢脱氮过程,基于DFT理论考察了吡啶和吡咯在α-Mo2C(0001)表面的吸附,结果显示吡啶和吡咯以大π键的形式吸附在3个Mo原子形成的三角型的位置。
笔者所在课题组前期研究表明,各种石化及煤焦油柴油馏分中咔唑类衍生物含量较高[10],且在咔唑、喹啉、吲哚、吡啶、苯胺等5种含氮化合物中,咔唑在MoS2表面的吸附能最大[11]。因此笔者选择咔唑及含量较高的2种咔唑类化合物3-甲基咔唑、1,5-二甲基咔唑,用Materials Studio 7.0构建晶体结构后,基于DFT理论采用Vienna ab initio Simulation Package(VASP)软件包进行了咔唑类化合物在MoS2表面吸附过程的优化计算,再通过GAUSSIAN 03软件对吸附分子全局参量(高低占据轨道能量、电负性和亲电指数)的计算揭示了引起其吸附能力差异的原因,以期为研究含氮化合物对加氢脱硫反应影响的作用机理提供理论依据。
1 MoS2晶体模型和密度泛函计算方法
MoS2晶体模型取自Materials Studio 7.0的结构数据库,由Build模块建立MoS2(0 0 -3)表面,采用(4×4)超晶胞模拟,晶胞间真空层厚度大于1 nm,图1为优化前后咔唑在3层MoS2表面吸附示意图。采用DFT理论的VASP程序包进行了表面及整个吸附过程的结构优化,计算了咔唑类化合物分子在不同层数的MoS2晶体表面的吸附能。周期性边界条件的计算使用Projector-augmented Wave(PAW)结合PW91泛函,其中平面波基组的能量截断为450 eV。咔唑类化合物分子在晶体表面的吸附能△E通过式(1)计算。
△E=Et-Es-Em
(1)
式(1)中,Et为分子吸附晶体稳定体系的总能量,eV;Es为晶体晶簇结构的能量,eV;Em为自由吸附分子的能量,eV。△E为正,表明吸附作用不能自发进行,需要外加能量;△E为负,则表明吸附为放热过程,吸附过程可以自发进行,且△E绝对值越大,吸附越稳定。
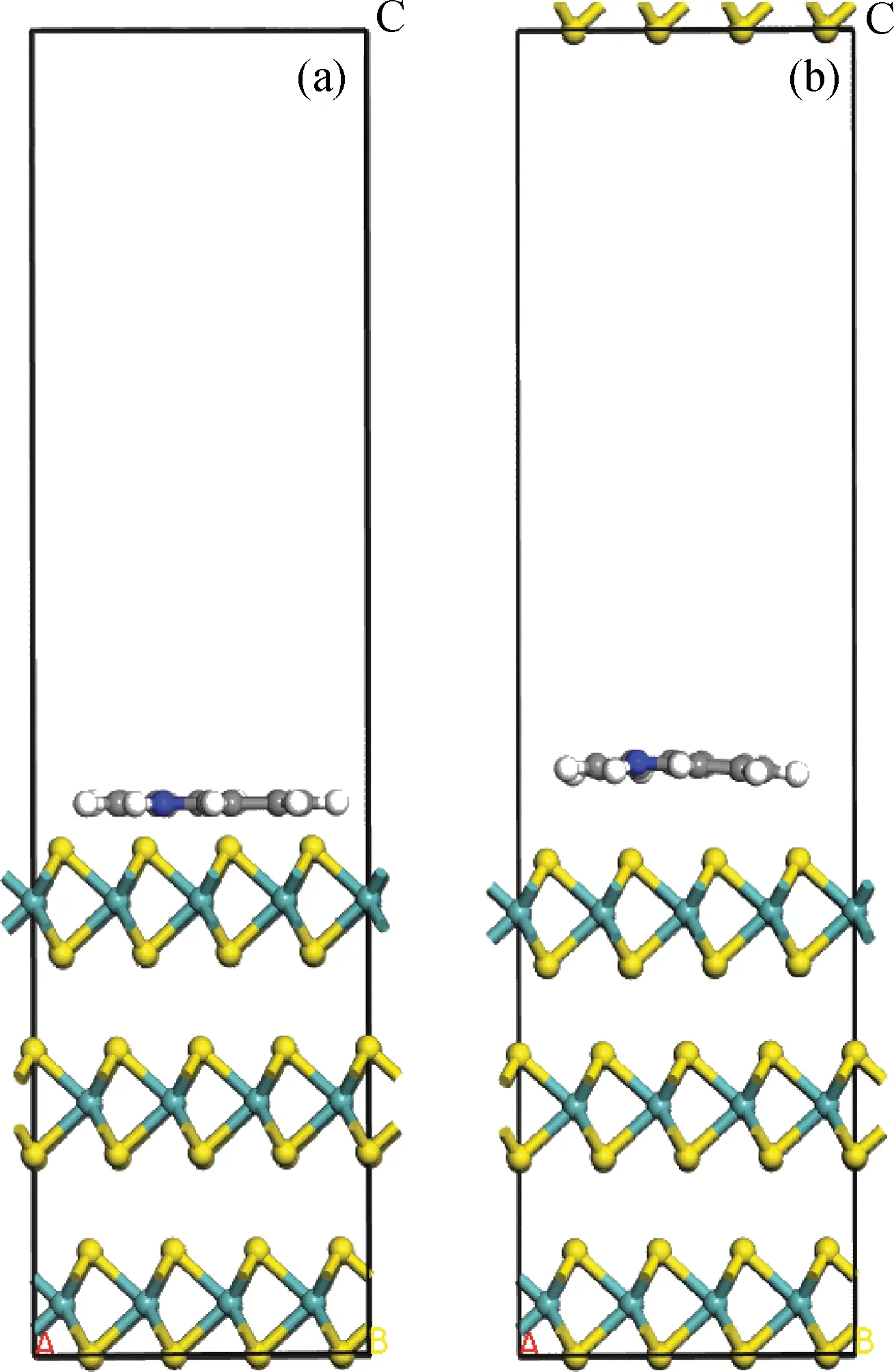
图1 咔唑在3层MoS2晶体表面吸附模型Fig.1 Structure model of carbazole on 3 slabs of MoS2 crystalBlue—Molybdenum atom; Yellow—Sulfur atomA, B and C are x, y and z axises.(a) Before optimization; (b) After optimization
采用GAUSSIAN 03软件包,用DFT理论中的杂化B3LYP方法,结合6-31G+(d,p)基组,计算了咔唑类化合物分子的最高占据轨道能量EHOMO和最低空轨道能量ELUMO,根据得到的EHOMO和ELUMO,通过公式(2)和(3)计算了咔唑类化合物分子的电负性χ和亲电指数ω[12-13]。
(2)
ω=(EHOMO+ELUMO)2/2(ELUMO-EHOMO)
(3)
2 结果与讨论
2.1 MoS2片层数对咔唑类化合物吸附能的影响
由文献[4]和笔者所在课题组前期所做工作可知,咔唑是以平行吸附形式吸附在单片层MoS2晶体表面的[11],笔者在此基础上考察了咔唑类化合物在不同片层数的MoS2催化剂上的平行吸附行为。咔唑类化合物均是含有1个N原子、甲基取代苯环不同位置的H原子形成的衍生物,具有大的π-共轭刚性平面结构。由图1可见,优化后咔唑分子整体从MoS2晶体表面向上移动,2个苯环距离催化剂表面较近,而咔唑的中心吡咯环距离催化剂表面较远,即苯环与MoS2表面的作用明显强于吡咯环,说明咔唑分子主要通过苯环吸附在MoS2晶体表面,这一结果与文献[4,6]所得的结论一致。
表1列出了咔唑分子在不同层数MoS2晶体表面吸附前后键长的变化以及吸附能。咔唑分子各原子编号如图2所示。由表1可见,固定1层MoS2时,咔唑分子在其表面吸附后的C—C、N—C键比其在真空中的键长略小;当MoS2层数增加到2时,由于底层原子固定,上层原子弛豫,使吸附后咔唑分子的键长显著减小,同时咔唑在2层MoS2表面的吸附能绝对值为1.73 eV,也显著小于其在1层MoS2表面的吸附能绝对值5.18 eV;当MoS2层数增加至3时,底层原子固定,上面两层原子弛豫,吸附后咔唑的键长略小于其在2层MoS2表面吸附后的键长,而咔唑在其表面的吸附能绝对值也相应略小于2层MoS2时的吸附能绝对值。这表明催化剂弛豫原子层数对吸附分子的结构影响较大,而固定层数的影响较小。随着弛豫原子层数增多,吸附后吸附分子的键长变短,键能变大,其与催化剂表面相互作用趋于稳定,从而使其在MoS2表面的吸附能也趋于恒定。考虑计算效率,笔者选择3层MoS2模型研究咔唑类化合物在其表面的吸附。赵巍等[14]计算了水分子在3层和4层Fe表面的吸附能,发现相对误差不超过0.08%,印证了计算的合理性。
咔唑、3-甲基咔唑和1,5-二甲基咔唑在3层MoS2晶体表面的吸附能分别为-1.30 eV、-1.24 eV、-0.93 eV,由文献[5]可知,单环含氮化合物吡啶和吡咯在NiMoS催化剂上不同吸附构型的吸附能分别在-1.29~-0.10 eV和-0.71~-0.68 eV范围内,比较可知笔者计算所得的吸附能结果较为合理。

图2 咔唑分子各原子编号示意图Fig.2 Schematic of the atomic number of carbazole molecule

表1 咔唑在不同层数MoS2上吸附前后的键长和吸附能Table 1 Bond distances in free carbazole and adsorbed carbazole on different slabs of MoS2
2.2 甲基取代基对咔唑类化合物吸附能的影响
随着咔唑类化合物苯环上甲基取代数目增多,其在MoS2晶体表面的吸附能由大到小的顺序为咔唑、3-甲基咔唑、1,5-二甲基咔唑。当咔唑分子处于自由状态时,由于N的吸电子作用,离N原子较远的C2—C3和C3—C4键长较长,键能较小。吸附后,咔唑类化合物的2个苯环优先吸附在MoS2表面,吡咯环凸起于催化剂表面,但吡咯环是1个刚性平面结构的五元环,由于分子张力的作用,C11—C12键长最长,键能最小。因此咔唑分子在束缚状态下,C11—C12键很可能先发生断裂。当苯环上有甲基取代基后,由于空间位阻效应阻碍了吸附分子上苯环在MoS2晶体表面的吸附,使得吸附能变小。文献[15]研究发现,二苯并噻吩(DBT)系列在NiMoS催化剂上平行吸附时,随着DBT分子上氢原子数目增多,减少了吸附分子与催化剂表面的相互作用点,使得吸附能随之减小。这一变化规律与本文研究结果类似。
计算了咔唑类化合物分子的最高占据轨道能EHOMO和最低非占据轨道能ELUMO,并由此得到咔唑类化合物分子的电负性χ和亲电指数ω,电负性和亲电指数是反映分子在反应中吸引电子能力强弱的参数。表2为咔唑类化合物的全局参量。由表2可知,作为供电子基团,随着咔唑类化合物苯环上甲基取代数目增多,分子的电负性和亲电指数有所减小,这可能是由于甲基的斥电子作用使得咔唑类化合物分子吸电子能力减弱,相应的,其在MoS2表面的吸附能随之变小,吸附越不稳定。这表明咔唑类化合物在MoS2晶体表面的吸附过程中为电子受体,同时也从另一角度阐述了甲基取代基对吸附能的影响。综上所述,随着咔唑类化合物分子中苯环上甲基取代数的增加,由于空间位阻和电子效应的共同作用,使其在MoS2晶体表面的吸附能变小。

表2 咔唑类化合物的全局参量Table 2 Part of global parameters of carbazole compounds
2.3 电子结构对咔唑类化合物吸附能的影响
为了深入考察咔唑类化合物分子在MoS2表面的吸附能与原子间电荷转移的关系,采用Bader电荷分析方法,通过在一个区域中整合电子密度从而量化了原子间电荷的转移情况。表3列出了咔唑类化合物分子中C原子、N原子的Bader电荷数和转移电荷数(净电荷),净电荷为正表明该原子失电子,净电荷为负表明该原子得电子。
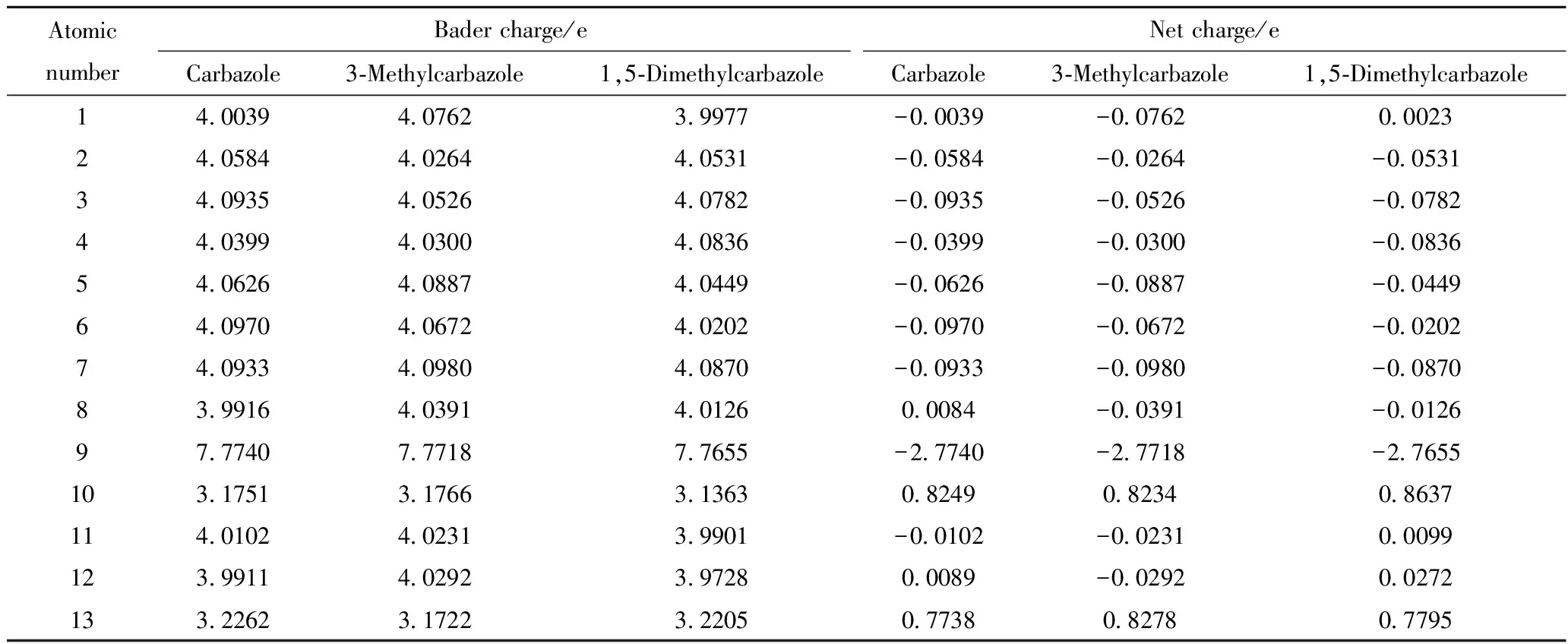
表3 咔唑类化合物的Bader电荷Table 3 Bader charges of carbazole compounds
从表3可以看出,咔唑类化合物分子中电荷转移比较显著的位置是N原子和与其相邻的2个C原子(10号C原子和13号C原子)。3种咔唑类化合物分子上C、N原子的电子转移情况基本一致:N原子得电子,与其相邻的10位和13位的C原子失电子,其他位的C原子电子基本不发生转移。Sun等[16]认为,对于咔唑分子,其亲电反应的活性中心在N原子上,亲核反应的活性中心分布在苯环上,这与笔者的研究结果相吻合。3种分子的N原子得电子由多到少的顺序为咔唑、3-甲基咔唑、1,5-二甲基咔唑,与它们在MoS2晶体表面的吸附能大小顺序一致,这一结果与Shiraishi等[17]研究的甲基对咔唑中N原子电子密度的变化规律类似。这进一步证明了咔唑类化合物在与MoS2催化剂吸附过程中为电子受体。
图3为咔唑类化合物分子的最高占有轨道(HOMO)和最低未占轨道(LUMO)的0.02等值面图。由图3可以看出,3种分子的 HOMO 轨道有大致相同的分布中心,对应的吡咯环内的π成键轨道分布一致,并且有N原子的p轨道贡献。3种分子的LUMO轨道主要离域在吡咯环上,是典型的π反键轨道分布。这种分布使得咔唑类化合物分子N原子上的孤对电子转移至催化剂Mo原子的缺电子中心形成配位键,同时吸附分子利用其LUMO轨道接受来自催化剂S原子HOMO轨道的电子形成反馈键,有利于咔唑类化合物分子在MoS2晶体表面稳定吸附。
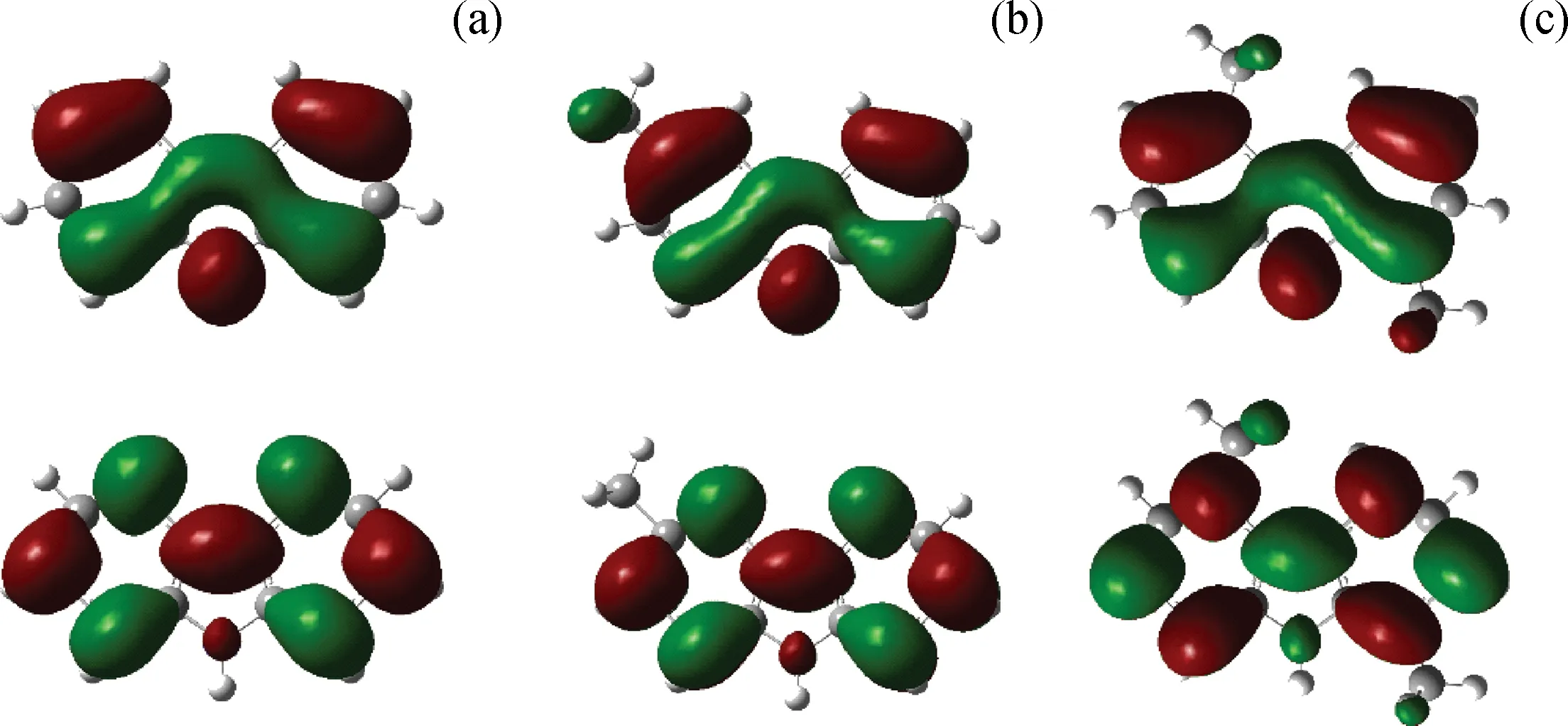
图3 咔唑类化合物分子的最高占有轨道(HOMO)和最低未占轨道(LUMO)的0.02等值面图Fig.3 The 0.02 isosurface figure of carbazole compounds molecular HOMO and LUMO The above three pictures are HOMO orbits and the following are LUMO orbits.(a) Carbazole; (b) 3-Methylcarbazole; (c) 1,5-Dimethylcarbazole
3 结 论
咔唑类化合物(咔唑、3-甲基咔唑和1,5-二甲基咔唑)在MoS2(0 0 -3)晶体表面的平行吸附能随着MoS2晶体片层数增多而减弱。由于空间位阻和电子效应的共同作用,使得咔唑类化合物在MoS2晶体表面的吸附能随着苯环上甲基取代数目的增加而变小。咔唑类化合物在MoS2晶体表面的吸附过程中为电子受体,吡咯环N上的电子流向催化剂Mo原子,同时咔唑类化合物分子的LUMO轨道接受S原子HOMO轨道的电子形成反馈键,使得咔唑类化合物在MoS2晶体表面稳定吸附。
[1] 邵志才, 高晓冬, 李皓光, 等. 氮化物对柴油深度和超深度加氢脱硫的影响I氮化物含量的影响[J].石油学报(石油加工), 2006, 22(4): 12-17. (SHAO Zhicai, GAO Xiaodong, LI Haoguang, et al. Effect of nitrogen-containing compounds on deep and ultradeep HDS for diesel oilⅠEffect of the content of nitrogen-containing compounds[J].Acta Petrolei Sinica (Petroleum Processing Section), 2006, 22(4): 12-17.)
[2] 张生, 李翔, 王安杰, 等. 有机含氮化合物对深度加氢脱硫反应的影响[J].工业催化, 2005, 13(11): 12-17. (ZHANG Sheng, LI Xiang, WANG Anjie, et al. Influence of organic nitrogen compounds on deep hydrodesulfurization(HDS)[J].Industrial Catalysis, 2005, 13(11): 12-17.)
[3] SHIRAISHI Y, TACHIBANA K, HIRAI T, et al. A novel desulfurization process for fuel oils based on the formation and subsequent precipitation of S-alkylsulfonium salts. 5. Denitrogenation reactivity of basic and neutral nitrogen compounds[J].Industrial & Engineering Chemistry Research, 2001, 40(22): 4919-4924.
[4] SUN M, NELSON A E, ADJAYE J. First principles study of heavy oil organonitrogen adsorption on NiMoS hydrotreating catalysts[J].Catalysis Today, 2005, 109(1): 49-53.
[5] SUN M, NELSON A E, ADJAYE J. Adsorption and hydrogenation of pyridine and pyrrole on NiMoS: An ab initio density-functional theory study[J].Journal of Catalysis, 2005, 231(1): 223-231.
[6] 孙炜, 方元, 王鹏, 等. 有机氮化物在镍钼硫催化剂的表面吸附[J].武汉工程大学学报, 2013, 35(12): 35-39. (SUN Wei, FANG Yuan, WANG Peng, et al. Adsorption of organonitrogen on surface of nickel-molybdenum sulfide catalyst[J].Journal of Wuhan Institute of Technology, 2013, 35(12): 35-39.)
[7] ABDALLAH W A, NELSON A E. Density functional theory study of pyrole adsorption on Mo(110)[J].The Journal of Physical Chemistry B, 2005, 109(21): 10863-10870.
[8] TEMEL B, TUXEN A K, KIBSGAARD J, et al. Atomic-scale insight into the origin of pyridine inhibition of MoS2-based hydrotreating catalysts[J].Journal of Catalysis, 2010, 271(2): 280-289.
[9] REN J, WANG J, HUO C F, et al. Adsorption of NO, NO2, pyridine and pyrrole onα-Mo2C(0001): A DFT study[J].Surface Science, 2007, 601(6): 1599-1607.
[10] 杨敬一, 周秀欢, 蔡海军, 等. 煤焦油和石油基柴油馏分中含氮化合物的分离鉴定[J].石油炼制与化工, 2015, 46(7): 107-112. (YANG Jingyi, ZHOU Xiuhuan, CAI Haijun, et al. Separation and identification of nitrogen compounds in diesel fraction of coal tar and petroleum[J].Petroleum Processing and Petrochemicals, 2015, 46(7): 107-112.)
[11] 杨敬一, 周秀欢, 袁佩青, 等. 几种含氮化合物在MoS2晶面的吸附研究[J].计算机与应用化学, 2015, 32(7): 769-773. (YANG Jingyi, ZHOU Xiuhuan, YUAN Peiqing, et al. The adsorption of nitrogen compounds on MoS2[J].Computers and Applied Chemistry, 2015, 32(7): 769-773.)
[12] GEERLINGS P, DE PROFT F, LANGENAEKER W. Conceptual density functional theory[J].Chemical Reviews, 2003, 103(5): 1793-1874.
[13] CHERMETTE H. Chemical reactivity indexes in density functional theory[J].Journal of Computational Chemistry, 1999, 20(1): 129-154.
[14] 赵巍, 汪家道, 刘峰斌, 等. H2O分子在Fe(100), Fe(110), Fe(111)表面吸附的第一性原理研究[J].物理学报, 2009, 58(5): 3352-3358. (ZHAO Wei, WANG Jiadao, LIU Fengbin, et al. First principles study of H2O molecule adsorption on Fe(100), Fe(110) and Fe(111) surfaces[J].Acta Physica Sinica, 2009, 58(5): 3352-3358.)
[15] 孙炜, 王鹏, 杨犁, 等. 二苯并噻吩加氢脱硫过程在镍钼硫催化剂表面的吸附[J].武汉工程大学学报, 2013, 35(2): 51-56. (SUN Wei, WANG Peng, YANG Li, et al. Adsorption of dibenzothiophene hydrodesulfurization process over NiMoS catalyst[J].Journal of Wuhan Institute of Technology, 2013, 35(2): 51-56.)
[16] SUN M, NELSON A E, ADJAYE J. Correlating the electronic properties and HDN reactivities of organonitrogen compounds: An ab initio DFT study[J].Journal of Molecular Catalysis A: Chemical, 2004, 222(1): 243-251.
[17] RAYBAUD P, HAFNER J, KRESSE G, et al. Structure, energetics, and electronic properties of the surface of a promoted MoS2catalyst: An ab initio local density functional study[J].Journal of Catalysis, 2000, 190(1): 128-143.
First Principles of the Adsorption of Carbazole Compounds on MoS2
YANG Jingyi, HE Xiao, ZHOU Xiuhuan, YUAN Peiqing, XU Xinru
(ChemicalEngineeringCollege,EastChinaUniversityofScienceandTechnology,Shanghai200237,China)
The adsorption behavior and adsorption energy of three carbazole compounds (carbazole, 3-methylcarbazole, 1,5-dimethylcarbazole) on MoS2cluster were investigated by the VASP package based on the Density functional theory (DFT). The results showed that carbazole compounds were electron acceptors in the process of adsorption on the surface of MoS2and the adsorption energy of carbazole compounds on MoS2reduced along with growing amount of MoS2slabs. As the amount of methyl substitution on benzene ring increased, the adsorption energy of carbazole compounds on MoS2reduced due to the steric hindrance of methyl and electronic effect. According to the Bader charge, HOMO and LUMO analysis, the adsorption energy of carbazole compounds on MoS2increased along with increased amount of electrons on N atom. It was observed that the lone pair electrons on N atom of adsorbed pyrrole ring are transferred to the electron-deficient center on Mo atoms, and the LUMO orbit of carbazole compounds accepts electrons in the HOMO orbit of S atoms, leading to the stable adsorption of carbazole compounds on the surface of MoS2.
carbazole compounds; MoS2cluster; adsorption; density functional theory; Bader charge
2016-07-25
国家自然科学基金项目(21376075)资助
杨敬一,男,副教授,博士,从事石油与能源化工方面的研究;E-mail:jyyang@ecust.edu.cn
1001-8719(2017)03-0515-06
TQ015.9
A
10.3969/j.issn.1001-8719.2017.03.016
