高效液相色谱法测定氨咖黄敏胶囊中对乙酰氨基酚含量
2016-04-01袁常珍李翅翔苟鹏英
胡 俊,袁常珍,廖 晴,李翅翔,苟鹏英
(泸州市食品药品检验所中药室,四川泸州 646000)
高效液相色谱法测定氨咖黄敏胶囊中对乙酰氨基酚含量
胡 俊,袁常珍,廖 晴,李翅翔,苟鹏英
(泸州市食品药品检验所中药室,四川泸州 646000)
目的:采用高效液相色谱法测定氨咖黄敏胶囊中对乙酰氨基酚的含量。方法:采用Hypersil ODS2(4.6 mm×200 mm,5 μm)色谱柱,以甲醇-0.05mol/L磷酸二氢钾溶液-三乙胺(20∶80∶0.02)(用磷酸调pH为3.2)为流动相,流速1 mL/min;检测波长243 nm;柱温30℃。结果:对乙酰氨基酚浓度在18.09~90.43 μg/mL范围内与峰面积呈良好的线性关系,r=0.9999,平均加样回收率为101.7%,RSD为0.58%。结论:此方法简便、准确、重现性好,可用于测定氨咖黄敏胶囊中对乙酰氨基酚含量。
高效液相色谱法;氨咖黄敏胶囊;对乙酰氨基酚
对乙酰氨基酚是常用解热镇痛药,过去一直认为该药是安全有效的。但近年来美国食品药品管理局(FDA)多次报道服用过量的对乙酰氨基酚可导致严重的肝损伤,甚至死亡[1]。因此准确控制高含量制剂中对乙酰氨基酚含量显得极为必要。氨咖黄敏胶囊是由对乙酰氨基酚、马来酸氯苯那敏、人工牛黄、咖啡因组成的复方制剂,其中对乙酰氨基酚的标示含量高达250 mg。因其能快速有效的缓解普通感冒和流行性感冒引起的发热、头痛、四肢酸痛等临床症状,成为深受患者青睐的常用非处方感冒药。其现行质量标准为国家食品药品监督管理局国家药品标准WS-10001-(HD-0276)-2002-2006。该标准采用容量分析法测定复方制剂中对乙酰氨基酚的含量,其前处理需要加稀盐酸加热回流,操作较为繁琐。滴定终点判定需用细玻璃棒蘸取溶液少许,划过涂有含锌碘化钾淀粉指示液的白瓷板上,观察条痕的颜色变化。这种终点判定的方法很不直观方便,既会因蘸取溶液而使成分损失又容易出现滴过终点的情况。亚硝酸钠滴定法的专属性也不高,应用于复方制剂的分析很容易受到其他药物成分和辅料成分的干扰。因此该含量测定方法的专属性、准确度、精密度和可操作性都有待进一步提高。鉴于此,笔者在查阅大量资料的基础上做了大量的摸索实验,最终选用专属性高的高效液相色谱法测定该复方制剂中对乙酰氨基酚的含量,取得了满意的效果,为该制剂质量检验提供了准确可靠的检测分析方法。
1 仪器与方法
1.1 仪器
Agilent 1200型高效液相色谱仪,包括四元梯度泵、真空脱气机、柱温箱、二极管阵列检测器、自动进样器、色谱工作站 (美国安捷伦科技有限公司);XS205DU十万分之一电子分析天平 (瑞士Mettler Toledo);Seven Excellence型酸度计 (瑞士 Mettler Toledo);101A-1BY电热恒温干燥箱(上海康路仪器设备有限公司);KH7200B型超声波清洗器(昆山禾创超声仪器有限公司);优普超纯水制造系统(成都超纯科技有限公司)。
1.2 材料与试药
三批氨咖黄敏胶囊(山西好医生药业有限公司,批号150 101,重庆迪康长江制药有限公司,批号151 101,重庆申高生化制药股份有限公司,批号151 202);对乙酰氨基酚对照品由中国食品药品检定研究院提供,编号100 018-201 409,纯度99.9%;咖啡因对照品由中国食品药品检定研究院提供,编号171 215-201 211,纯度99.9%;马来酸氯苯那敏对照品由中国食品药品检定研究院提供,编号100 047-201 507;人工牛黄对照药材由中国食品药品检定研究院提供,编号121 197-200 903;甲醇(进口色谱纯,Fisher Scientific公司);水为超纯水;其他试剂均为分析纯。
1.3 方法
1.3.1 色谱条件
色谱柱:伊利特Hypersil ODS2(4.6 mm×200 mm,5 μm);流动相:甲醇-0.05 mol/L磷酸二氢钾溶液-三乙胺(20∶80∶0.02)(用磷酸调pH为3.2);流速:1 mL/min;检测波长243 nm;柱温:30℃;进样量:10 μL(所有样品进样前过0.45 μm的滤膜)。
1.3.2 溶液配制
1.3.2.1 对照品溶液的制备
精密称取105℃干燥2 h的对乙酰氨基酚对照品适量,加25%甲醇溶液溶解并稀释成每1 mL含对乙酰氨基酚0.028 mg的溶液,作为对照品溶液。
1.3.2.2 供试品溶液的制备
取氨咖黄敏胶囊10粒的内容物混匀精密称取适量(约相当于对乙酰氨基酚0.07 g)置100 mL量瓶中,加25%甲醇溶液适量,超声振荡处理30 min,放冷,用25%甲醇溶液稀释至刻度,摇匀,滤过,精密量取续滤液2 mL置50 mL量瓶中,加25%甲醇溶液至刻度,摇匀,即得供试品溶液。
2 结 果
2.1 方法验证
2.1.1 系统适用性试验
取供试品溶液和对照品溶液按“1.3.1”色谱条件下方法进样,记录色谱图,结果见图1。结果表明理论塔板数按对乙酰氨基酚峰计算不低于11 000,对乙酰氨基酚峰与相邻峰分离度均大于4。
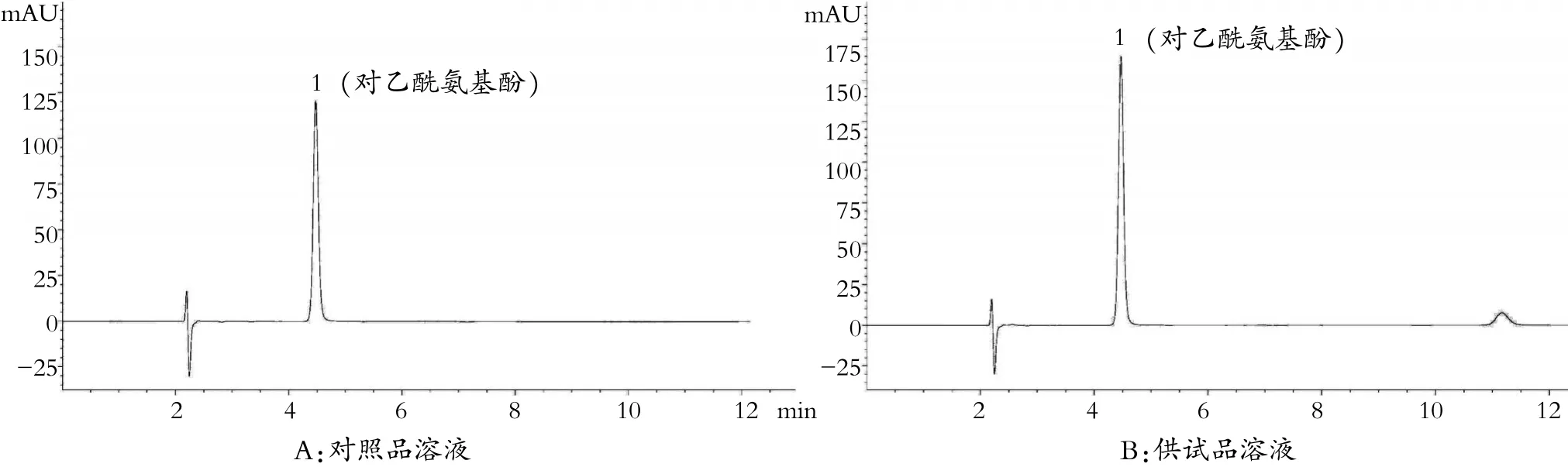
图1 系统适用性试验HPLC色谱图
2.1.2 专属性试验
取咖啡因、马来酸氯苯那敏和人工牛黄按氨咖黄敏胶囊处方比例制成缺对乙酰氨基酚阴性样品,照1.3.2.2供试品溶液制备方法制成缺对乙酰氨基酚阴性对照溶液,按“1.3.1”色谱条件下方法进样,记录色谱图,结果见图2。结果表明在对乙酰氨基酚保留时间处无干扰色谱峰,本方法的专属性良好。
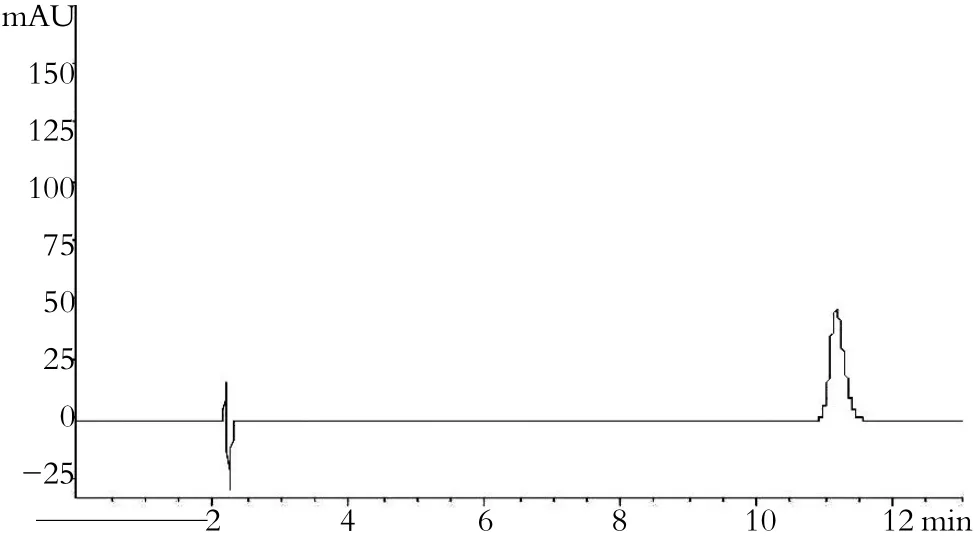
图2 专属性试验HPLC色谱图
2.1.3 线性关系考察
精密称取105℃干燥2 h的对乙酰氨基酚对照品45.26 mg,置100 mL量瓶中,用25%甲醇溶液溶解并稀释至刻度,摇匀,分别精密量取2 mL、4 mL、6 mL、8 mL、10 mL置50 mL量瓶中,加25%甲醇溶液稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,分别进样10 μL,记录色谱图,得到各浓度的峰面积值,结果见表1。以色谱峰面积值为纵坐标Y,以对乙酰氨基酚浓度(μg/mL)为横坐标X,绘制标准曲线,得曲线方程Y=32.5907X+0.7313,r=0.9999。结果表明对乙酰氨基酚浓度在18.09~90.43 μg/mL范围内与色谱峰面积值呈良好的线性关系。

表1 对乙酰氨基酚浓度与对应的色谱峰面积值
2.1.4 精密度试验
将同一对照品溶液,在相同的色谱条件下连续进样6次,每次10 μL,6次峰面积值见表2,RSD为0.53%,表明对乙酰氨基酚进样的精密度良好。

表2 6次进样的峰面积值
2.1.5 稳定性试验
将同一供试品溶液在室温下分别放置0、2、4、8、12和24 h后进样,峰面积的RSD为0.61%,表明24 h内对乙酰氨基酚稳定性良好。
2.1.6 重复性试验
取同一生产批号(山西好医生药业有限公司,批号150 101)的样品,按上述供试品溶液制备方法和色谱条件进行6份平行测定,6份含量测定结果见表3,RSD为0.59%,表明本法测定的重复性良好。

表3 6份含量测定结果
2.1.7 加样回收率试验
照1.3.2.2供试品溶液的制备方法,精密称取已知含量的同一生产批号(山西好医生药业有限公司,批号150 101)的样品6份,精密加入浓度为0.5237 mg/mL的对照品溶液20 mL,制成供试品加标溶液进样分析,计算回收率,结果见表4,平均回收率为101.7%,RSD为1.3%,回收率符合有关规定。

表4 6份回收率
2.2 样品测定
取批号为150 101、151 101、151 202的三批氨咖黄敏胶囊分别制备供试品溶液,按“1.3.1”色谱条件下方法进样分析,记录色谱峰面积,按外标法以峰面积计算,测得对乙酰氨基酚含量分别为标示量的99.7%、99.3%、99.5%。三批样品用容量分析法测得结果分别为99.4%、98.9%、99.2%,相对偏差分别为0.15%、0.20%、0.15%。
3 讨论
3.1 检测方法类型的比较与选择
文献报道对乙酰氨基酚的常用含量测定方法有亚硝酸钠滴定分析法[2]、紫外分光光度法和高效液相色谱法[3]。亚硝酸钠滴定分析法存在缺陷,其前处理繁琐、复杂,外指示剂的使用进一步增加了操作环节,而且使得显色不够明显,终点不易掌握,人为的差异很大,需要经验,例如若溶液蘸取次数过多,容易造成损失,使结果判断不准确。通过对2.2样品测定结果进行对比分析,发现三批样品的亚硝酸钠滴定分析法测得数据均小于高效液相色谱法测得数据,这一结果正好应证了滴定分析法中蘸取溶液的确会造成损失导致测定结果偏小。亚硝酸钠滴定法的专属性也不高,应用于复方制剂的分析很容易受到其他药物成分和辅料成分的干扰[4]。紫外分光光度法的专属性同样不高,用于复方制剂的含量测定同样会受到其它药物成分和辅料成分的干扰。因此本品作为复方制剂适宜选用专属性高、操作方便的高效液相色谱法进行含量测定。
3.2 流动相的选择
通过对流动相:甲醇-0.05 mol/L磷酸二氢钾溶液-三乙胺(20∶80∶0.02)(用磷酸调pH为3.2)、甲醇-水-磷酸(22∶78∶0.1)[3]、甲醇-0.5%冰醋酸溶液(20:80)[5]和0.05 mol/L醋酸铵溶液-甲醇(85∶15)[6]进行实验比较,结果甲醇-0.05 mol/L磷酸二氢钾溶液-三乙胺 (20∶80∶0.02)(用磷酸调pH为3.2)系统分离效果和拖尾因子良好,保留时间适度,故选择本系统为流动相。
3.3 检测波长的确定
通过二极管阵列检测器对对照品溶液在200~400 nm进行扫描,结果在243 nm处有最大吸收,故选择243 nm作为检测波长。
3.4 溶解溶剂的选择
文献报道溶解对乙酰氨基酚的溶剂有25%甲醇溶液、水、氢氧化钠溶液。经实验比较对乙酰氨基酚在25%甲醇溶液中溶解性良好,在溶液中成分的结构形态稳定且该溶液与流动相的匹配性良好,故选择25%甲醇溶液作为溶解溶剂。
3.5 供试品溶液制备提取方式的选择
供试品提取方式有振摇、加热回流和超声振荡三种常用方式。加热回流操作较繁琐且会使被测成分对乙酰氨基酚不稳定;振摇溶解速度较慢且占用人工效率低下;超声振荡不仅操作方便快捷而且还有被测成分稳定的优点。所以选择超声振荡提取制备供试品溶液。
3.6 供试品溶液制备提取时间的确定
试验发现超声振荡15~60 min试验结果基本一致,为确保对乙酰氨基酚充分溶解选择超声振荡30 min。
3.7 小结
实验结果显示用本方法测定对乙酰氨基酚含量,样品预处理简单,系统适用性优良,阴性对照无干扰专属性强,线性、精密度、稳定性、重复性、准确度均良好,更适用于对乙酰氨基酚的含量控制。
1.司继刚,赵群,曹原.对乙酰氨基酚肝毒性评价分析[J].医学综述,2015,21(19):3537-3538.
2.王玉.药品检验指南(上册)[M].北京:中国医药科技出版社,2011:293-294.
3.国家药典委员会.中华人民共和国药典[S].第二部.北京:中国医药科技出版社,2010:234-238.
4.王慧文,张士洋.氨咖黄敏胶囊中对乙酰氨基酚含量3种测定方法的比较[J].安徽医药,2005,9(5):354-356.
5.国家药典委员会.中华人民共和国药典[S].第二部.北京:中国医药科技出版社,2010:1141.
6.董超琪.HPLC法测定对乙酰氨基酚滴剂的含量[J].海峡药学,2014,26(5):52-53.
(2016-01-20收稿)
Determination of paracetamol in Ankahuangmin capsule by HPLC
Hu Jun,Yuan Changzhen,Liao Qing,Li Chixiang,Gou Pengying
TCM chanber of Food and Drug Inspertion,Luzhou 646000,Sichuan Province,China
Objective:To determine the content of paracetamol in Ankahuangmin capsule by HPLC.Methods:Hypersil ODS2 (4.6 mm×200 mm,5 μm)column,and the moving phase of methanol-0.05 mol/L potassium dihydrogen phosphate solution-triethylamine (20∶80∶0.02,pH 3.2 adjusted with phosphoric acid)were used in the experiment.The flow rate was 1 mL/min,detection wavelength 243 nm,and column temperature was 30℃.Results:Paracetamol concentration in the range of 18.09~90.43 μg/mL and the peak area was linear relationship,r=0.9999,the average recovery was 101.7%,RSD was 0.58%.Conclusion:This method is simple, accurate,reproducible,and can be used to determine the content of paracetamol in Ankahuangmin capsule.
HPLC;Ankahuangmin capsule;Paracetamol
R917
A
10.3969/j.issn.1000-2669.2016.02.013
胡 俊(1974-),男,主管药师。E-mail:1048225901@qq.com
