氧化应激诱导自噬对骨髓间充质干细胞增殖与凋亡的影响*
2016-01-31刘关羽何卫阳黄小龙印胡滨
刘关羽, 何卫阳, 朱 鑫, 杨 帆, 黄小龙, 印胡滨, 苟 欣
(1重庆医科大学附属第一医院泌尿外科,2分子肿瘤及表观遗传学重庆市重点实验室,重庆 400016)
氧化应激诱导自噬对骨髓间充质干细胞增殖与凋亡的影响*
刘关羽1, 2,何卫阳1△,朱鑫1,杨帆1,黄小龙1, 2,印胡滨1, 2,苟欣1
(1重庆医科大学附属第一医院泌尿外科,2分子肿瘤及表观遗传学重庆市重点实验室,重庆 400016)
[摘要]目的: 观察氧化应激是否可诱导骨髓间充质干细胞(MSCs)自噬,探索在氧化应激微环境中自噬对MSCs增殖和凋亡的影响及可能机制,以期提高移植MSCs治疗糖尿病勃起功能障碍(DMED)的效果。方法: 以过氧化氢(H2O2)模拟氧化应激微环境。台盼兰染色细胞计数法及MTT分析检测不同浓度(0 、50、100、200、400 μmol/L)H2O2对MSCs增殖的影响。联合MTT、流式细胞术、Western blot及透射电镜等方法研究H2O2对MSCs凋亡及自噬的影响。结果: H2O2呈浓度依耐性抑制MSCs增殖,IC50=(384.5751±16.8895)μmol/L;Western blot分析及透射电镜提示H2O2在诱导MSCs凋亡的同时,诱导MSCs产生自噬;MTT分析及流式细胞术提示H2O2组细胞增殖受抑、凋亡率上升,阻断自噬后细胞增殖进一步减弱,凋亡率进一步上升;Western blot结果提示H2O2诱导cleaved caspase-3增多及多聚ADP-核糖聚合酶1(PARP1)裂解增强;阻断凋亡后,cleaved caspase-3降低及PARP1裂解减弱;阻断自噬后,cleaved caspase-3进一步增多及PARP1裂解进一步增强。结论: 氧化应激对MSCs存活具有双重作用,在诱导MSCs凋亡的同时还诱导保护性自噬,阻断MSCs自噬可以增加MSCs的凋亡,因此进一步增强细胞保护性自噬将有望提高移植MSCs在糖尿病高氧化应激微环境中的存活率,从而改善移植MSCs对糖尿病勃起功能障碍的疗效。
[关键词]自噬; 间充质干细胞; 细胞增殖; 细胞凋亡; 氧化应激
据报道,截至2030年,全球糖尿病患者将达3.36亿,约半数糖尿病患者同时伴有勃起功能障碍(erectiledysfunction,ED)[1]。糖尿病病人患ED比非糖尿病人早10~15年,其ED严重程度也更高,严重影响着患者的家庭生活以及社会和谐[2]。在糖尿病高糖状态下,氧化应激微环境损害阴茎海绵体神经和内皮细胞,神经元型一氧化氮合酶(neuronalnitricoxidesynthase,nNOS)和内皮型一氧化氮合酶(endothelialnitricoxidesynthase,eNOS)活性降低,一氧化氮释放减少,这是糖尿病勃起功能障碍(diabetesmellituserectiledysfunction,DMED)的主要发病机制之一[3]。目前治疗ED的一线药物主要是磷酸二酯酶抑制剂(phosphodiesteraseinhibitors5,PDEI5),但其对DMED却疗效甚微[4]。
骨髓间充质干细胞(mesenchymalstemcells,MSCs)因在具体微环境中可定向分化为阴茎海绵体血管内皮细胞和平滑肌细胞而被用于治疗DMED的研究[5],但前期研究表明,MSCs移植后对勃起功能改善作用持续7~28d后就逐渐消失[6-7],推测可能与移植的MSCs无法适应糖尿病患者阴茎海绵体内高氧化应激微环境而出现异常细胞凋亡有关[8-9]。因此,如何提高移植MSCs存活率成为亟待解决的问题。
细胞自噬是一个降解细胞内受损蛋白及细胞器并为细胞增殖提供再利用原料的复杂过程,对MSCs自我更新、分化及存活起非常重要的调节作用[10]。一定程度的自噬对细胞存活起保护作用,但过度自噬又会导致细胞死亡,自噬和凋亡的关系复杂,通过多个细胞信号系统相互影响[11-12]。本实验拟通过调节MSCs自噬水平,观察在氧化应激作用下自噬对MSCs增殖和凋亡的影响,为改善MSCs移植治疗DMED提供理论依据。
材料和方法
1主要试剂和仪器
小鼠骨髓间充质干细胞株C3H10购于ATCC;DMEM/F12培养基购自HyClone;胎牛血清(fetalbovineserum,FBS)购自Gibco;青霉素、链霉素购自北京碧云天生物技术研究所;3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐[3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazoliumbromide,MTT]为Amresco分装产品;氟聚偏乙烯(polyvinylidenefluoride,PVDF)膜、增强型化学发光液(enhancedchemiluminescence,ECL)购自Millipore;预染蛋白Marker购自Thermo; 自噬抑制剂氯喹(chloroquine,CQ)、3-甲基腺嘌呤(3-methyladenine,3-MA)及微管相关蛋白1轻链3(microtubule-associatedprotein1lightchain3,LC3)I抗购自Sigma;caspase抑制剂z-VAD购自Selleck;cleaved-caspase-3I抗购自CST;P62及多聚ADP-核糖聚合酶1(poly-ADP-ribosepolymerase1,PARP1)I抗购自BD;β-actinI抗购自钟鼎公司;II抗购自Abgen。倒置相差显微镜(Olympus);凝胶成像仪(FusionFX);流式细胞仪(BectonDickinson);透射显微镜(HITACHI)。
2主要方法
2.1细胞培养MSCs培养在含10%FBS和1%青、链霉素的DMEM/F12培养基中,置于37 ℃、5%CO2、饱和湿度培养箱中常规传代培养。取对数生长期细胞用于实验。
2.2细胞计数法检测细胞增殖变化收集对数生长期细胞按1×104/cm2接种于6孔板,待细胞生长至70%~80%融合时加入不同浓度(0、50、100、200、400 μmol/L)H2O2。分别向各孔加入对应浓度H2O2,处理细胞24 h后胰酶消化下各孔细胞并用台盼兰染色计数。
2.3MTT实验检测细胞活力的变化根据H2O2浓度不同将细胞分成 0、50、100、200、400 μmol/L 共5组;根据加入抑制剂不同,将细胞分为 对照组(常规培养)、H2O2组、H2O2+ z-VAD组、 H2O2+3-MA组、H2O2+CQ组及H2O2+CQ+z-VAD组,共6组,其中3-MA为5 mmol/L ,CQ为10 μmol/L,z-VAD为20 μmol/L,各抑制剂均预处理细胞30 min。收集对数生长期的MSCs,按每孔1×104接种于96孔板培养至细胞融合度达70%~80%,按上述分组加入H2O2处理细胞24h,换液,加入5g/LMTT试剂作用4h,弃上清,洗涤,再加入二甲基亚砜(dimethylsulfoxide,DMSO)溶解紫蓝色结晶物甲瓒,于570nm处测吸光值(A)。每组设3复孔,重复实验3次,计算各组细胞活力抑制率。抑制率=(1-A实验组/A对照组)×100%。
2.4Westernblot实验MSCs培养至70%~80%融合,H2O2处理细胞不同时间(针对凋亡相关蛋白cleavedcaspase-3及PARP1处理0、2、4、8h;针对自噬相关蛋白LC3、P62处理0、1、2、4、8h)后提取细胞总蛋白,BCA法测蛋白浓度后蛋白变性,按40μg/孔上样进行SDS-PAGE分离蛋白,再将蛋白转印至PVDF膜,脱脂奶粉常温封闭1~2h,I抗4 °C过夜,洗膜,II抗常温孵育1~2h,再洗膜,ECL发光液显影。“自噬潮”实验分为对照组(常规培养)、CQ处理细胞2.5h组、H2O2处理细胞2h组、H2O2+CQ合用药组,其中合用药组先用CQ预处理细胞0.5h,再加入H2O2继续处理2h,提取细胞总蛋白检测LC3表达变化。根据加入抑制剂不同分组如下:对照组(常规培养)、H2O2处理细胞8h组、H2O2+z-VAD合用药组、H2O2+CQ合用药组、H2O2+CQ+z-VAD合用药组,各合用药组均先用抑制剂(z-VAD、CQ)预处理细胞0.5h,再加入H2O2继续处理8h,提取细胞总蛋白检测cleavedcaspase-3表达及PARP1裂解变化。以上各组中H2O2浓度均为300μmol/L,CQ均为10μmol/L,z-VAD均为20μmol/L。
2.5透射扫描电镜检测细胞自噬体实验分为正常组(常规培养)和H2O2(300μmol/L)处理细胞 4h组。MSCs培养至70%~80%融合,按上述分组处理细胞后收集细胞并用PBS洗涤1~2次,离心后弃上清,加入2.5%戊二醛4 ℃固定,送样检验细胞自噬体。
2.6流式细胞术AnnexinV/PI双标法检测细胞凋亡以下各组中H2O2浓度为300μmol/L,CQ均为10μmol/L,z-VAD均为20μmol/L。分为对照组(未做任何处理)、H2O2处理组、H2O2+z-VAD合用药组、H2O2+CQ合用药组、H2O2+CQ+z-VAD合用药组,各合用药组均先用抑制剂(z-VAD、CQ)预处理细胞 0.5h,再加入H2O2继续处理8h。将细胞培养至70%~80%融合,按上述分组处理细胞后,消化收集所有培养液及细胞,按AnnexinV/PI凋亡检测试剂盒说明操作,1h内上机检测细胞凋亡。
4统计学处理
用SPSS19.0统计软件进行分析。数据采用均数±标准差(mean±SD)表示,多组间比较采用单因素方差分析(one-wayANOVA),组间两两比较使用Bonferroni-t检验,以P<0.05为差异有统计学意义。
结果
1MSCs增殖抑制率随H2O2浓度增加逐渐上升
用不同浓度(0、50、100、200、400μmol/L)H2O2处理细胞24h,台盼兰计数及MTT法检测细胞增殖抑制率,发现随H2O2浓度的上升,MSCs增殖抑制率逐渐上升,IC50=(384.5751±16.8895)μmol/L,选择300μmol/L作为后续实验的基础浓度,见图1。
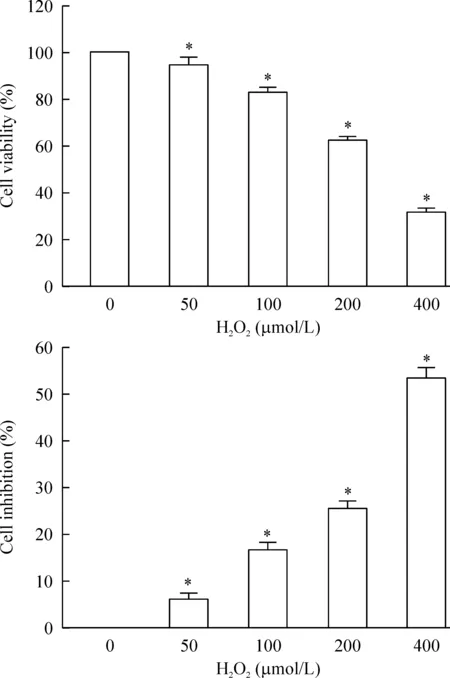
Figure1.TheeffectsofH2O2atdifferentconcentrationsoncellviabilityandproliferationofMSCsafter24h.Mean±SD. n=3.*P<0.05vs0 μmol/L group.
图1不同浓度H2O2处理MSCs24h对其存活及增殖的影响
2H2O2在诱导MSCs凋亡的同时,诱导MSCs自噬
用H2O2(300μmol/L)处理细胞不同时间(0、2、4、8h),Westernblot检测发现随时间推移,cleavedcaspase-3的蛋白水平及PARP1裂解逐渐增强。用H2O2处理细胞不同时间(0、1、2、4、8h)发现,随时间推移,LC3-Ⅱ 表达逐渐上调,P62表达逐渐下调,提示H2O2诱导MSCs发生自噬。为进一步明确LC3-Ⅱ 表达上调是自噬引起而非自噬底物降解受阻造成的结果,本实验进一步检测“自噬潮”。H2O2组及CQ组LC3-Ⅱ 表达较正常组有一定程度上调;而H2O2+CQ组LC3-Ⅱ表达较H2O2组又进一步上调,表明LC3-Ⅱ的上调是H2O2诱导的自噬而非自噬底物降解受阻造成的结果。透射电镜结果显示,与正常组对比,H2O2处理细胞4h后引起MSCs自噬体明显增加。以上结果提示H2O2在诱导MSCs发生凋亡的同时诱导MSCs发生自噬,见图2。
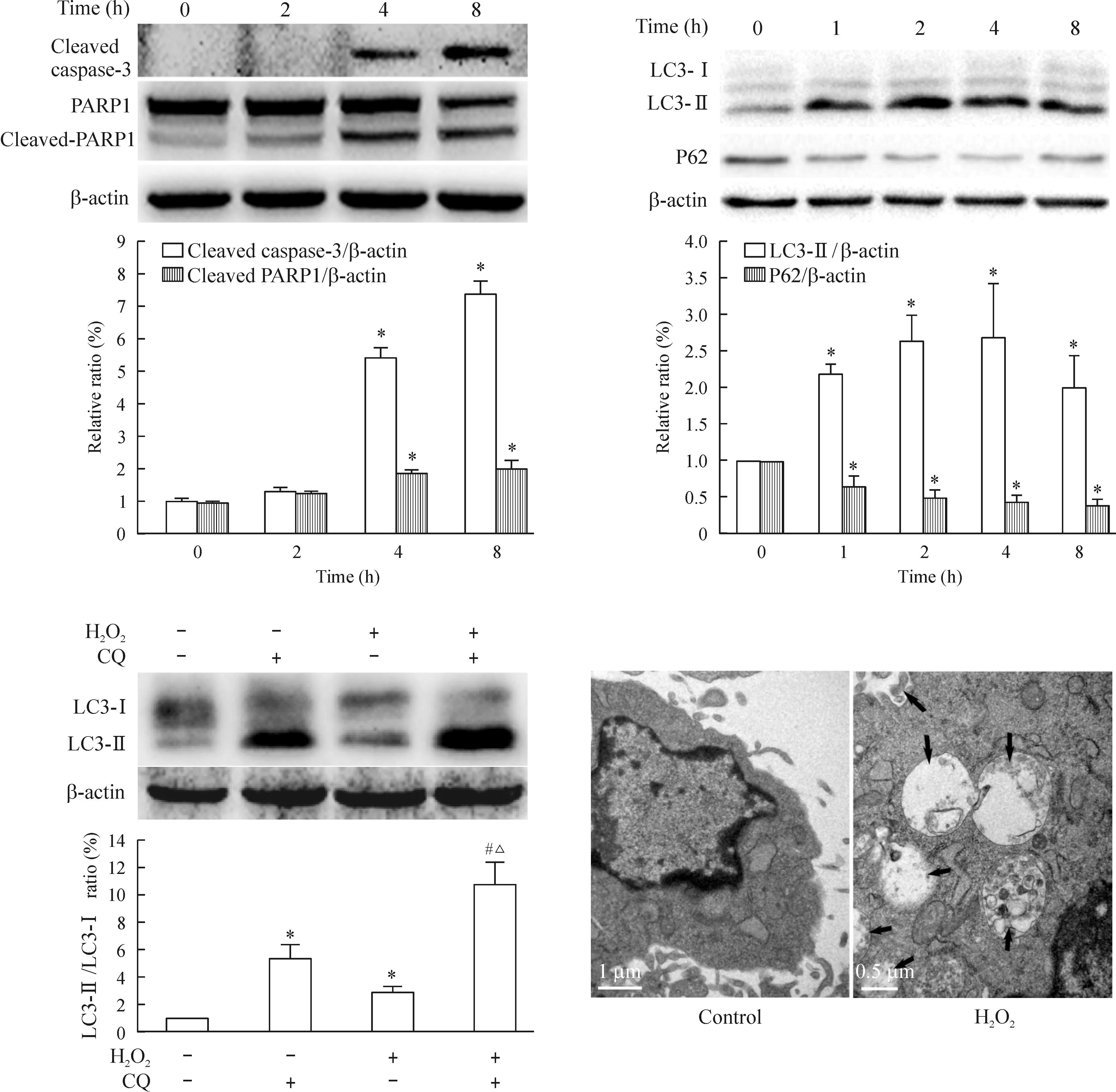
Figure2.BothapoptosisandautophagywereinducedbyH2O2.Theblackarrowindicatedtheautophagybodycontainingdigestiveendproducts.Mean±SD. n=3.*P<0.05vscontrol group;#P<0.05 vsCQgroup;△P<0.05vsH2O2group.
图2H2O2在诱导MSCs发生凋亡的同时,诱导MSCs发生自噬
3氧化应激微环境下,MSCs发生自噬对细胞起保护作用
MTT实验结果说明,H2O2组的细胞活力抑制率较正常组明显上升,H2O2+z-VAD组的细胞活力抑制率较H2O2组明显下降,H2O2+3-MA组和H2O2+CQ组的细胞活力抑制率较H2O2组进一步上升,而H2O2+CQ+z-VAD组的细胞活力抑制率较H2O2+CQ组又有所下降(P<0.05),见图3。此外,H2o2组的细胞凋亡率较正常组明显上升,H2O2+z-VAD组的细胞凋亡率较H2O2组明显下降,H2O2+CQ组的细胞凋亡率较H2O2组进一步上升,而H2O2+CQ+z-VAD组的凋亡率较H2O2+CQ组又有所下降(P<0.05)。Westernblot技术检测cleavedcaspase-3的蛋白水平及PARP1的裂解变化显示H2O2组cleavedcaspase-3的蛋白水平及PARP1的裂解较正常组明显上调;H2O2+z-VAD组cleavedcaspase-3的蛋白水平及PARP1的裂解较H2O2组明显下调,H2O2+CQ组cleavedcaspase-3的蛋白水平及PARP1的裂解较H2O2组进一步上调,而H2O2+CQ+z-VAD组cleavedcaspase-3的蛋白水平及PARP1的裂解较H2O2+CQ组又有所下调(P<0.05)。以上均提示H2O2诱导MSCs发生自噬对细胞存活起保护作用,细胞死亡是通过细胞凋亡(Ⅰ型程序性死亡)途径而非过度自噬(Ⅱ型程序性死亡)途径发生的,见图4。
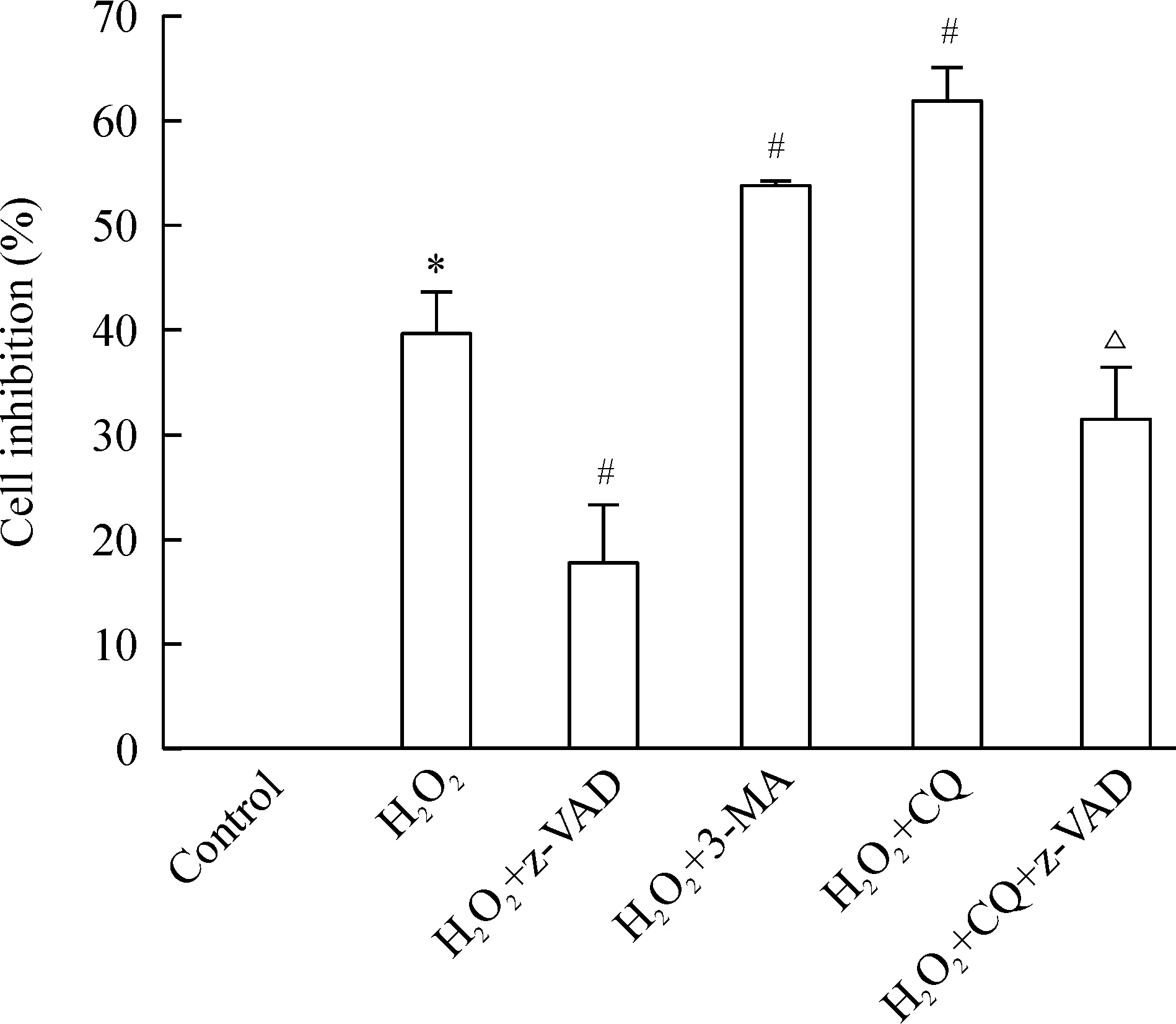
Figure3.TheeffectsofautophagicandapoptosisblockadecombinewithH2O2onthecellactivityofMSCsfor24h.Mean±SD. n=3.*P<0.05vscontrol group;#P<0.05 vsH2O2group;△P<0.05vsH2O2+CQ group.
图3H2O2联合自噬及凋亡抑制剂处理MSCs24h对细胞活力的影响
讨论
与非糖尿病人相比,糖尿病患者其勃起功能障碍发病更早、患病率更高、程度也更严重,严重影响着患者的生活质量[1-2]。目前治疗ED的一线药物主要是PDEI5(如:西地那非、伐地那非、他达那非等),但这类药物可以引起一系列不良反应(如:头痛、发热、消化不良、腹泻等),且它对糖尿病性ED的疗效远低于非糖尿病患者[4]。
干细胞具有“高度增殖、自我更新和多向分化”的特性,在特定微环境下可定向分化为阴茎海绵体血管内皮细胞和平滑肌细胞[5],因此人们希望通过移植MSCs来修复受损的阴茎血管内皮细胞和平滑肌细胞而到达治疗ED的目的。无论是功能学测定还是组织学评价都显示干细胞移植能够改善勃起功能,但该作用仅维持7~28d后就逐渐消失,其原因与移植的干细胞无法适应受体的微环境而出现细胞过度凋亡有关[6-7]。因此,如何提高移植干细胞在受体内的存活率,是目前ED干细胞治疗亟待解决的关键问题。
糖尿病高血糖导致机体处于高氧化应激状态,这种状态会损害阴茎海绵体神经和内皮细胞,nNOS和eNOS的活性降低,NO释放减少,这是糖尿病ED的主要发病机制之一[3]。在链脲霉素诱导的糖尿病勃起功能障碍大鼠模型中,其阴茎海绵体内氧化应激产物如丙二醛等大量聚集,而抗氧化物质超氧化物歧化酶和谷胱甘肽含量明显下降,不能有效清除ROS,导致糖尿病ED阴茎海绵体组织中ROS水平增加[14-15]。研究显示,即使应用胰岛素将血糖控制在正常范围,因为线粒体的“代谢记忆”,即糖化的线粒体继续产生过量ROS,这种高氧化应激状态依然存在[16]。研究表明,ROS会影响骨髓间充质干细胞自我更新、分化,加速干细胞和内皮祖细胞衰老,增加其凋亡,降低受损血管的自我修复能力[9]。
细胞自噬是一个降解细胞内受损的细胞器和蛋白质并为细胞增殖提供再利用原料的复杂过程,可在细胞处于内外界压力(如氧化压力、低氧、营养缺乏等环境)时帮助细胞度过难关,同时自噬对干细胞的自我更新及分化发挥重要的调节作用[17]。
研究表明氧化应激在诱导细胞凋亡的同时促进细胞自噬[18],但氧化应激环境下自噬与凋亡的关系比较复杂,关于在糖尿病患者阴茎海绵体高氧化应激微环境中移植MSCs是否发生自噬、自噬与细胞存活及凋亡的关系,目前国内外尚未见文献报道。本实验用H2O2模拟高氧化应激微环境,探讨体外MSCs在高氧化应激环境中凋亡及自噬水平的变化,并通过调节MSCs自噬水平,观察其在氧化应激微环境下对MSCs增殖和凋亡的影响。首先,台盼兰计数法及MTT实验测得细胞增殖率随H2O2浓度上升逐渐下降。以酶原形式存在于胞浆中的caspase-3被激活生成活性片段cleavedcaspase-3以及PARP1 裂解被看作是细胞发生凋亡的标志之一。LC3存在胞浆可溶型LC3-Ⅰ和膜结合型LC3-Ⅱ 2种形式,当自噬被激活时,LC3-Ⅱ 随自噬体膜的增多而增多,是自噬体的标志分子,因此LC3-Ⅱ 表达上调可作为细胞发生自噬的标志之一。P62是一个泛素结合蛋白,参与自噬过程并被降解,其表达下调可反映自噬活性增强[13]。本实验发现,随时间推移,LC3-Ⅱ、cleavedcaspase-3和cleavedPARP1的蛋白水平逐渐上调而P62逐渐下调,说明氧化应激微环境下细胞在发生凋亡的同时诱导自噬。“自噬潮”及透射电子显微镜实验进一步证明氧化应激可诱导MSCs发生自噬。为探讨在氧化应激微环境下细胞发生的自噬与凋亡的关系,本实验结合MTT及流式细胞术发现H2O2抑制细胞增殖,增加细胞凋亡;阻断自噬后细胞增殖进一步受抑制,凋亡率进一步上升,而凋亡抑制剂z-VAD能反转这一结果。

Figure4.TheprotectiveeffectsofautophagyinducedbyH2O2ontheMSCs.Mean±SD. n=3.*P<0.05vscontrol group;#P<0.05 vsH2O2group;△P<0.05vsH2O2+CQ group.
图4H2O2诱导MSCs自噬对细胞起保护作用
Westernblot实验发现类似结果,H2O2能增加凋亡相关蛋白cleavedcaspase-3及cleavedPARP1的水平;阻断自噬后,cleavedcaspase-3及cleavedPARP1进一步上调;而阻断凋亡后,cleavedcaspase-3及cleavedPARP1明显下调。以上结果均提示氧化应激引起MSCs死亡是通过凋亡途径而非过渡自噬途径实现的,其诱导的自噬对细胞起保护作用,但氧化应激诱导MSCs发生自噬的具体机制仍需要进一步探索。
综上所述,氧化应激对MSCs存活具有双重作用,在促进MSCs凋亡的同时还诱导保护性自噬,阻断MSCs自噬可以显著增加MSCs凋亡,因此进一步增强细胞保护性自噬可能有望提高移植MSCs在糖尿病高氧化应激微环境的存活率,从而改善移植MSCs对糖尿病勃起功能障碍的疗效。本实验的成功实施,将为移植MSCs治疗DMED提供新的思路。
[参考文献]
[1]PhéV,RouprêtM.Erectiledysfunctionanddiabetes:Areviewofthecurrentevidence-basedmedicineandasynthesisofthemainavailabletherapies[J].DiabetesMetab, 2012, 38(1):1-13.
[2]MariaMI,BellastellaG,EspositoK.Diabetesandsexualdysfunction:currentperspectives[J].DiabetesMetabSyndrObes, 2014, 7:95-105.
[3]BurnettAL,LowensteinCJ,BredtDS,etal.Nitricoxide:aphysiologicmediatorofpenileerection[J].Science,1992, 257(5608):401-403.
[4]MalavigeLS,LewJC.Erectiledysfunctionindiabetesmellitus[J].JSexMed, 2009, 6(5):1232-1247.
[5]AlwaalA,ZaidUB,LinCS,etal.Stemcelltreatmentoferectiledysfunction[J].AdvDrugDelivRev, 2014, 21(12):1280-1285.
[6]FandelTM,AlbersenM,LiuG,etal.Recruitmentofintracavernouslyinjectedadipose-derivedstemcellstothemajorpelvicganglionimproveserectilefunctioninaratmodelofcavernousnerveinjury[J].EurUrol, 2012, 61(1): 201-210.
[7]GouX,HeWY,XiaoMZ,etal.TransplantationofendothelialprogenitorcellstransfectedwithVEGF165torestoreerectilefunctionindiabeticrats[J].AsianJAndrol, 2011,13(2):332-338.
[8]MottaqhiS,LarijaniB,SharifiAM.Apelin13:anovelapproachtoenhanceefficacyofhypoxicpreconditionedmesenchymalstemcellsforcelltherapyofdiabetes[J].MedHypotheses, 2012, 79(6):717-718.
[9]YaoEH,YuY,FukudaN.Oxidativestressonprogenitorandstemcellsincardiovasculardiseases[J].CurrPharmBiotechnol, 2006, 7(2):101-108.
[10]MortensenM,WatsonAS,SimonAK.Lackofautophagyinthehematopoieticsystemleadstolossofhematopoieticstemcellfunctionanddysregulatedmyeloidproliferation[J].Autophagy, 2011, 7(9):1069-1070.
[11]ChenW,SunY,LiuK,etal.Autophagy:adouble-edgedswordforneuronalsurvivalaftercerebralischemia[J].NeuralRegenRes, 2014, 9(12):1210-1216.
[12]胡鹏飞,赖东武,何红. 自噬在晚期糖基化终产物诱导的内皮细胞凋亡中的作用[J]. 中国病理生理杂志,2012, 28(6):1006-1011.
[13]秦正红, 乐卫东.《自噬——生物学与疾病》[M]. 第1版. 北京:科学出版社,2011:55-57.
[14]LongT,LiuG,WangY,etal.TNF-α,erectiledysfunction,andNADPHoxidase-mediatedROSgenerationincorpuscavernosuminhigh-fatdiet/streptozotocin-induceddiabeticrats[J].JSexMed, 2012, 9(7):1801-1814.
[15]LiM,ZhuanL,WangT,etal.ApocyninimproveserectilefunctionindiabeticratsthroughregulationofNADPHoxidaseexpression[J].JSexMed, 2012, 9(12):3041-3050.
[16]IhnatMA,ThorpeJE,KamatCD,etal.Reactiveoxygenspeciesmediateacellular“memory”ofhighglucosestresssignalling[J].Diabetologia, 2007, 50(7):1523-1531.
[17]GuanJL,SimonAK,PrescottM,etal.Autophagyinstemcells[J].Autophagy, 2013, 9(6):830-849.
[18]KaminskyyVO,ZhivotovskyB.Freeradicalsincrosstalkbetweenautophagyandapoptosis[J].AntioxidRedoxSignal, 2014, 21(1):86-102.
(责任编辑: 林白霜, 余小慧)

*[基金项目]国家自然科学基金资助项目(No. 81200254);山西省回国留学人员科研基金资助项目(No. 2014-033);山西省研究生创新基金资助项目(No. 20133075)
Effectofoxidativestress-inducedautophagyonproliferationandapoptosisofMSCsLIUGuan-yu1, 2,HEWei-yang1, ZHU Xin1,YANGFan1, HUANG Xiao-long1, 2, YIN Hu-bin1, 2, GOU Xin1
(1DepartmentofUrinarySurgery,TheFirstAffiliatedHospitalofChongqingMedicalUniversity,2Chongqing Key Laboratory of Molecular Oncology and Epigenetics, Chongqing 400016, China. E-mail: gouxincq@163.com)
[ABSTRACT]AIM: To investigate whether oxidative stress is able to induce autophagy in mesenchymal stem cells (MSCs), and to explore the effects of autophagy on MSC proliferation and apoptosis under oxidative stress circumstance as well as the underlying mechanism for promoting the therapeutic effects of transplanted MSCs on treating diabetes mellitus erectile dysfunction (DMED). METHODS: Hydrogen peroxide (H2O2) was applied to simulate the oxidative stress circumstance. The effects of H2O2at concentration of 0, 50, 100, 200, 400 μmol/L on the viability of MSCs were tested by the method of Trypan blue exclusion and MTT assay respectively . The methods of MTT assay, Western blot and transmission electron microscope (TEM) were used to explore the effects of H2O2on MSC apoptosis and autophagy. RESULTS: The proliferation of MSCs was obviously inhibited by H2O2in a dose-dependent manner (P<0.01) and the 50% inhibiting concentration (IC50) was (384.58±16.89) μmol/L. H2O2induced apoptosis and autophay of MSCs. The proliferation rate of MSCs was suppressed by H2O2significantly (P<0.05), with a further decline by blockade of autophagy (P<0.05) whereas increased by blockade of apoptosis (P<0.05). H2O2induced MSCs apoptosis obviously (P<0.05), with an augment of apoptosis (P<0.05) by blockade of autophagy. Furthermore, the H2O2increased expression of cleaved caspase-3 and cleavage of poly ADP-ribose polymerase 1 (PARP1), Which were decreased by apoptosis blockade whereas were enhanced by blockade of autopahgy. CONCLUSION: Oxidative stress plays a dual role in MSC survival, which induces MSC apoptosis and autophagy. Moreover, blockade of autophagy intensifies MSC apoptosis. Therefore, it is a promising method to ameliorate the effects of stem-cell based therapy on DMED by enhancing protective autophagy to increase the survival rate of transplanted MSCs against oxidative stress circumstance caused by diabetes mellitus.
[KEY WORDS]Autophagy; Mesenchymal stem cells; Cell proliferation; Apoptosis; Oxidative stress
通讯作者△Tel: 0351-4135787; E-mail: luli7300@126.com
[收稿日期]2015- 04- 30[修回日期] 2015- 09- 07
[文章编号]1000- 4718(2015)12- 2183- 05
doi:10.3969/j.issn.1000- 4718.2015.12.011
[中图分类号]R363.2; R698+.1
[文献标志码]A
