超高效液相色谱串联质谱法测定银耳中米酵菌酸
2015-07-23周鹏福建省产品质量检验研究院福建福州350002
周鹏(福建省产品质量检验研究院,福建福州350002)
超高效液相色谱串联质谱法测定银耳中米酵菌酸
周鹏
(福建省产品质量检验研究院,福建福州350002)
摘要:建立了快速测定银耳中米酵菌酸的超高效液相色谱串联质谱分析方法。样品采用甲醇提取,混合强阴离子交换固相萃取柱(PAX)净化,UPLCBEH C18(2.1×100mm,1.7μm)色谱柱分析,以10mmol/L乙酸铵-乙腈为流动相,梯度洗脱,串联四级杆质谱多反应监测负离子模式检测,基质匹配外标法定量。结果表明,米酵菌酸在1.6μg/L~80μg/L线性范围内,相关系数(r)0.999 9,检出限0.5μg/kg,加标回收率82%~102%,相对标准偏差RSD≤8.5%。本方法快捷准确、灵敏度高,可用于银耳中米酵菌酸的确证方法。
关键词:超高效液相色谱-串联质谱;米酵菌酸;银耳;固相萃取;基质效应
米酵菌酸(bongkrekic acid,BKA)是椰毒假单胞菌酵米面亚种代谢物中的一种,它存在于发酵食品、变质银耳等食物中,毒性较强,食用者会出现恶心、呕吐、抽搐、休克等中毒症状,损伤肝、脑神经细胞以及肾脏组织[1-2],致死率高达40%~100%,在我国引起多起食物中毒事件,需严加防范。近期研究又发现米酵菌酸对细胞线粒体通透性转换孔有抑制作用,从而又开始对米酵菌酸的生物合成及其在细胞凋亡、损伤等方面展开了大量的研究[3-8]。在我国最新颁布的相关标准中[9-10]明确规定了银耳中米酵菌酸含量小于0.25mg/kg。目前米酵菌酸的检测方法较少,主要有液相色谱法[11]、薄层色谱法[11]以及紫外分光光度法[12],液质联用分析法还鲜见报道。现有方法处理复杂,灵敏度差、易受到样品基质的干扰,使得测定结果出现偏差。根据我国相关规定:基于色谱分析而没有使用分子光谱测定的方法,不能用于确证方法[13]。因此,急需开发一种处理快速、特异性强、灵敏度高的分析方法,作为银耳中米酵菌酸的确证方法。
1 材料与方法
1.1仪器、试剂与材料
1290超高效液相色谱仪串联6490三重四级杆质谱仪:均为美国Agilent公司;离心机:Avanti;J-E冷冻高速离心机:美国贝克曼公司;DS-8510DTH超声波清洗机:上海弗鲁克流体机械制造有限公司;VORTEXMAX II型涡旋混合器:美国Thermo Scientific公司;固相萃取仪:美国安捷伦科技公司。
1.0mg/m L米酵菌酸标准品:Sigma公司;甲醇、乙腈、甲酸(均为色谱纯):美国Merck公司;乙酸铵(优级纯):北京百灵威科技公司;氢氧化钠、氨水、磷酸(均为分析纯):国药集团;试验用水:milli-Q制备的超纯水;混合强阴离子交换固相萃取柱(PAX,60mg/3mL):艾杰尔公司。
1.2标准溶液的配制
准确移取0.50mL标准品溶液于25mL容量瓶中,甲醇定容,配制成20mg/L的标准储备液。分别移取适量的标准储备液用50%甲醇溶液稀释定容,配制成系列标准工作溶液,4℃下避光保存。
1.3液相色谱条件
1.3.1色谱条件
色谱柱:Waters BEH C18>2.1×100mm>1.7μm;柱温:40℃;进样量:2.0μL;流速:0.3m L/min;流动相A:0.01mol/L乙酸铵,流动相B:乙腈。梯度洗脱程序为:0~4min,20%B-40%B;4.01min~6min,90%B;6.01min~8.0min,20%B。
1.3.2质谱条件
离子源:电喷雾离子源(ESI);扫描方式:负离子模式;监测模式:多反应监测(MRM);脱溶剂气温度:160℃;脱溶剂气流速:16L/min;雾化气压力:241.3kPa;鞘气温度:350℃;鞘气流速:12 L/min;毛细管电压:3 000 V;喷嘴电压:0 V。监测离子对为485.1/441.1、485.1/397.2,定量离子对为485.1/441.1。
1.4样品前处理
样品经高速粉碎机粉碎后,准确称取2.0g于50mL具塞离心管中,加入10mL甲醇涡旋混匀,超声辅助提取30min。4000 r/min离心5min,准确移取5mL上层清液于5mL刻度氮吹管中,40℃下氮吹至干,加入3mL 0.1mol/LNaOH涡旋复溶后过PAX柱(固相萃取柱使用前分别用3mL甲醇、3mL 0.1mol/LNaOH活化,保持柱体湿润),再分别用3mL1%氨水(体积分数)、3mL1%氨水甲醇(体积分数)溶液淋洗,弃去淋洗液,抽干,最后用3mL 0.5%甲酸甲醇(体积分数)洗脱,洗脱液于45℃下氮吹至近干,用50%甲醇水(体积分数)溶液定容至1.0mL,过0.22μm尼龙滤膜后上机测试。
2 结果与讨论
2.1样品前处理条件优化
现有的米酵菌酸处理方法为液液萃取法[9-10],采用固相萃取法分析米酵菌酸还鲜见报道,需进一步优化。试验采用2.0mg/L米酵菌酸标准溶液,分别考察了亲水亲脂固相萃取柱(PEP,60mg/3mL)、混合弱阴离子交换柱(PWAX,60mg/3mL)、混合强阴离子交换柱(PAX,60mg/3mL)以及中性氧化铝柱(500mg/3mL)的保留特性。米酵菌酸经PEP、PWAX、中性氧化铝柱处理后,回收率为0%,而经PAX柱处理后,回收率可达到95%以上。分析发现,米酵菌酸极性较强,在PEP柱上无法保留,并且在PWAX柱上也无法保留,而在中性氧化铝柱上的保留又过强,难以洗脱;米酵菌酸在PAX柱上具有良好的保留特性,洗脱容易,回收率接近100%,适合米酵菌酸的富集、净化。试验发现,pH对米酵菌酸的洗脱强度有很大影响,考察了洗脱液中不同浓度甲酸对洗脱强度的影响。结果表明:当洗脱液中不含甲酸时几乎无法洗脱米酵菌酸,当甲酸浓度大于0.2%以后洗脱完全,且随着甲酸浓度的增大,回收率几乎无变化,试验结果见图1。
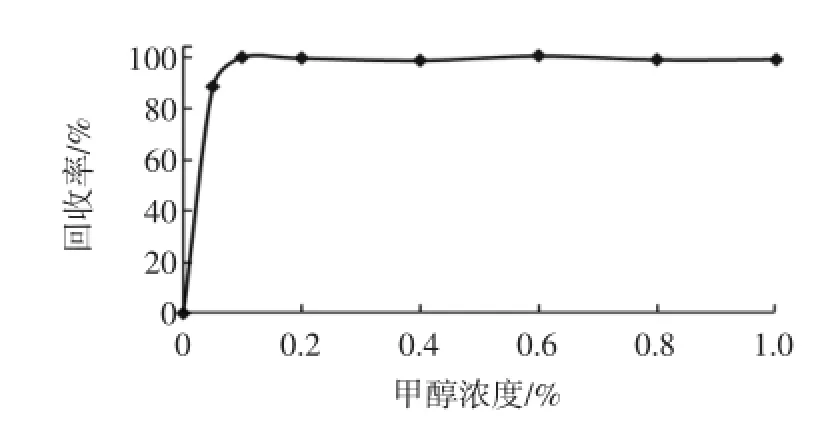
图1 不同甲酸浓度对洗脱强度的影响Fig.1 Effect of different concentration of form ic acid on the elution strength
考虑到基质的影响以及溶液配制的稳定性,最终选择了0.5%甲酸甲醇溶液作为洗脱剂。
2.2色谱质谱条件优化与基质效应
试验比较了乙腈-水、乙腈-0.3%甲酸、乙腈-0.01mol/L乙酸铵作为流动相对色谱峰型以及灵敏度的影响。由于米酵菌酸有3个羧基,具有一定极性,采用乙腈-水体系色谱峰变形严重,难以识别;采用乙腈-0.3%甲酸可以获得较好的色谱峰型,但是本试验采用的电喷雾质谱负离子模式(ESI-)进行检测,抑制了米酵菌酸的电离,灵敏度较低;采用乙腈-0.01mol/L乙酸铵体系可以获得较好的峰形以及较高的灵敏度。因此,本试验最终采用乙腈-0.01mol/L乙酸铵体系作为流动相。
试验采用2.0mg/L的标准品溶液进样,不经色谱柱分离,直接进入三重四级杆质谱仪进行质谱参数的优化。米酵菌酸容易失去一个质子形成[M-H]-离子,因此采用负离子模式进行检测。试验过程中优化了鞘气温度及流量、雾化气压力、去溶剂气温度及流速、毛细管电压、喷嘴电压、碰撞气能量以及碰撞池加速电压等参数,并选择[M-H]-离子(m/z485.1)为母离子。选定母离子后,进行子离子扫描,获得二级质谱图,见图2。
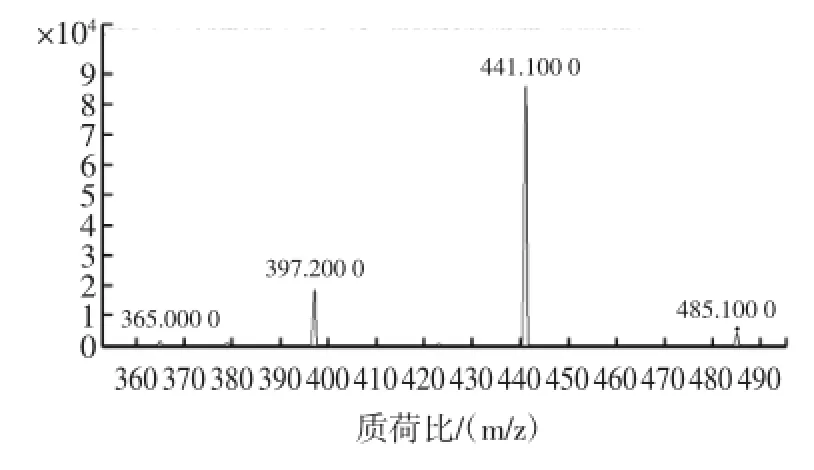
图2 米酵菌酸的二级质谱图Fig.2 Massspectrum of Bongkrekic acid by daughter scan
由图可见主要特征碎片为m/z441.1和m/z397.2,推测分别为[M-H-CO2]-和[M-H-2CO2]-峰。
2.3基质效应的探讨
不同于传统的荧光或紫外-可见光检测器,电喷雾质谱虽然对分离度的要求大为降低,基质不会影响到目标化合物的分离度,但是在其离子化过程中会产生基质效应,从而影响检测的准确度。通常降低基质效应的方法主要有稀释进样溶液、基质匹配标样以及稳定同位素内标法等,它们均有各自的优缺点,提高净化效率是最重要的途径[14-16],但是也很难完全消除基质效应的干扰。
试验采用浓度为40μg/L的基质匹配标样进行试验,发现通过调节梯度洗脱程序,米酵菌酸的峰面积变化很大,说明不同梯度洗脱条件下,米酵菌酸与基质的分离度差别很大,导致基质效应差异很大,最终选择了响应最大的为最佳洗脱条件,试验结果见图3。
图3 不同梯度洗脱程序米酵菌酸响应的影响Fig.3 Effectof differentgradientelution program on bongkrekic acid response
在优化后的洗脱程序下,分别采用浓度为40μg/L的纯溶剂配制标样与基质匹配标样,改变样品的进样量,分析米酵菌酸峰面积的变化,并进行数据拟合。理论上待测化合物的峰面积与进样量应为线性关系,但是基质匹配的标样在进样量小于2μL时,峰面积与纯溶剂配制的标样极为接近,即基质效应很小;随着进样量的增大,峰面积与溶剂配制的标样差别增大,即基质效应变强,对目标化合物的抑制越来越强;当进样量超过5μL时,进样量对目标化合物的响应影响有限。试验结果表明:进样量对基质效应的影响显著,采用液质联用法进行分析时,不能依靠加大进样量来提高方法的检出限。考虑到方法的灵敏度与基质效应,选择2μL为本试验的进样量,试验结果见图4。
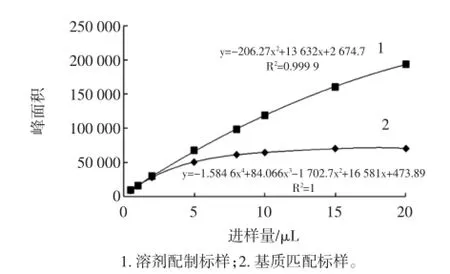
图4 不同进样量对米酵菌酸响应的影响Fig.4 Effectof different inject volumeon on bongk rekic acid response
2.4分析方法确证
在上述优化条件下,使用基质匹配标准曲线,外标法定量,考察了米酵菌酸在(1.6~80)μg/L范围内的线性关系和相关系数。以空白样品基质中米酵菌酸3倍信噪比,确定化合物的检出限为0.5μg/kg,方法的灵敏度能够满足检测要求。
对空白银耳样品在2.0、10、20μg/kg 3个加标水平下进行米酵菌酸的加标回收试验,考察方法的回收率和精密度,结果见表1。

表1 米酵菌酸的线性范围、线性方程、相关系数、回收率及精密度(n=6)Table1 Linear range,regression equations,cor relation coefficient,recoveriesand precision ofbongkrekic acid(n=6)
试验结果表面:在3个添加水平下,米酵菌酸的回收率为82%~102%,符合分析准确度要求;相对偏差为2.4%~8.5%,符合精密度要求。
3 结论
本研究建立了固相萃取净化-超高效液相色谱-串联质谱法检测银耳中米酵菌酸的方法。通过对分析方法的优化,采用基质匹配标准曲线的方法,建立了快速、准确、灵敏的超高效液相色谱-串联质谱法测定银耳中米酵菌酸的方法,同时本方法可作为分析的确证方法,为保障食品安全发挥积极作用。
参考文献:
[1]宋兴田,吴洪娟,庄宝祥.米酵菌酸中毒小鼠肝和脑组织的形态学改变[J].中国医药导报,2010,7(26):36-38
[2]宋兴田,吴洪娟,庄宝祥,等.米酵菌酸对小鼠肾组织毒性作用的超微结构观察[J].中国医学创新,2010,7(24):145-146
[3]Okuda K,HasuiK,AbeM,etal.Moleculardesign,synthesis,and evaluationof novel potent apoptosis inhibitors inspiredfrom bongkrekic acid[J].Chemical research in toxicology,2012,25(10): 2253-2260
[4]Rey M,ForestE,Pelosi L.Exploring the conformationaldynamicsof thebovine ADP/ATPcarrier inmitochondria[J].Biochemistry,2012, 51(48):9727-9735
[5]FrancaisA,Leyva A,Etxebarria-JardiG,etal.Totalsynthesisof the anti-apoptotic agents iso-and bongkrekic acids[J].Organic letters, 2009,12(2):340-343
[6]Zhao J,Zhou ZQ,Jin JC,etal.Mitochondrialdysfunction induced bydifferentconcentrationsofgadolinium ion[J].Chemosphere,2014, 100(4):194-199
[7]Moebius N,Ross C,Scherlach K,etal.Biosynthesisof the RespiratoryToxinBongkrekicAcidinthePathogenicBacterium Burkholderia gladioli[J].Chemistry&biology,2012,19(9):1164-1174
[8]Sato Y,Aso Y,Shindo M.Efficient synthesis of bongkrekic acid. Three-component convergentstrategy[J].Tetrahedron Letters,2009, 50(28):4164-4166
[9]中华人民共和国农业部.NY/T 749-2012绿色食品食用菌[S].北京:中国标准出版社,2012
[10]中华人民共和国国家卫生和计划生育委员会.GB 7096-2014食品安全国家标准食用菌及其制品[S].北京:中国标准出版社, 2014
[11]胡文娟,陈晓明,王玉华.酵米面、银耳、玉米中黄杆菌毒素A的薄层及高压液相色谱测定法[J].卫生研究,1986,15(2):31-34
[12]黄建立.银耳微波真空干燥机理及品质特性的研究[D].福州:福建农林大学,2010
[13]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.GB/T 27404-2008实验室质量控制规范食品理化检测[S].北京:中国标准出版社,2008
[14]Moragues F,Igualada C.How to decrease ion suppression in amultiresidue determination ofβ-agonists in animal liver and urine by liquid chromatography-massspectrometrywith ion-trap detector[J]. Analytica chimicaacta,2009,637(1):193-195
[15]Caban M,Migowska N,Stepnowski P,et al.Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination ofβ-blockersandβ-agonistsin environmentalsamples [J].Journalof Chromatography A,2012,1258:117-127
[16]周鹏,林钦,黄红霞,等.稳定同位素稀释超高效液相色谱-串联质谱法测定饲料中24种禁用兽药含量[J].分析化学,2014,42(2): 233-238
DOI:10.3969/j.issn.1005-6521.2015.22.031
收稿日期:2014-05-11
基金项目:福建省科技厅民生科技专项资助项目(2013Y6003)
作者简介:周鹏(1980—),男(汉),工程师,硕士,研究方向:复杂体系分离分析。
Determ ination of Bongkrekic Acid in Trem ella Fuciform is Berk by U ltra H igh Perform ance Liquid Chrom atography-tandem M ass Spectrometry
ZHOUPeng (Fujian Inspection and Research Institute for ProductQuality,Fuzhou 350002,Fujian,China)
Abstract:A method based on ultra high performance liquid chromatography coupled with tandem mass spectrometry(UHPLC-MS/MS)was developed for determination of bongkrekic acid(BAK)in tremella fuciformisberk.The samplewasextracted withmethanol,and then cleaned up by anion exchange solid-phase extraction(SPE).The targetanalytewasseparated on UPLCBEH C18(2.1×100mm,1.7μm)column,0.01mol/L ammonium acetate and acetonitrile as the mobile phase gradient elution,detected by MS/MS system under negative electrospray ionization(ESI-)modewithmultiple reaction ionmonitoring(MRM).The detection limit was 0.5μg/kg.The linear correlation coefficientwas 0.999 9.The recovery ranged from 82%to 102%with RSD less than 8.5%.Thismethod was suitable for the identification and quantification of bongkrekic acid in tremella fuciformisberk due to itssimplicity,good purification and high sensitivity.
Key words:ultra high performance liquid chromatography-tandem mass spectrometry(UHPLC-MS/MS);bongkrekic acid(BKA);tremella fuciformisberk;solid phase extraction(SPE);matrix effect
