高效液相色谱法测定牛黄清火丸中蒽醌类化合物的含量
2015-07-07刁全平郭华吕琳琳李铁纯侯冬岩
刁全平,郭华,吕琳琳,李铁纯,侯冬岩
(鞍山师范学院 化学与生命科学学院,辽宁 鞍山 114007)
高效液相色谱法测定牛黄清火丸中蒽醌类化合物的含量
刁全平,郭华,吕琳琳,李铁纯,侯冬岩Δ
(鞍山师范学院 化学与生命科学学院,辽宁 鞍山 114007)
目的 建立同时测定牛黄清火丸中大黄酸、大黄素、大黄酚、大黄素甲醚4种蒽醌类化合物含量的方法。方法 用20% 硫酸水解样品,以丙酮为溶剂,索氏提取法提取样品中的蒽醌类化合物,采用高效液相色谱法对其进行测定,色谱柱为 Kromasil C18柱,流动相为甲醇-0.5%磷酸水溶液(75:25,V/V),流速为1.0 mL/min,检测波长为230 nm。结果 样品中4种蒽醌类化合物分别在1.50~150、0.950~95.0、1.30~130、1.15~115 mg/L范围内呈良好的线性关系,4种化合物加标回收率在98.14%~101.3%之间。结论 该方法操作便捷,准确,稳定性好,可用于牛黄清火丸的质量控制。
牛黄清火丸;蒽醌类化合物;高效液相色谱法
牛黄清火丸是由大黄、黄芩、桔梗、牛黄、冰片、丁香、山药、雄黄、薄荷脑九味中药配伍而成的中成药,具有清热泻火、散风解毒等功效,主要用于治疗肝、胃、肺蕴热引起的头晕目眩、口鼻生疮、风火牙痛、咽喉肿痛等病症[1]。蒽醌类化合物具有较强的泻下、消炎、抗菌作用[2-4],是牛黄清火丸的主要功效成分,其含量的测定主要为HPLC法[5-10]。牛黄清火丸由于组分较多,成份复杂,在其产品质量的控制上有一定的难度。为了更好地控制该制剂质量,笔者通过对样品中蒽醌类化合物的提取、分离、测定方法的考查,建立了牛黄清火丸中大黄酸、大黄素、大黄酚、大黄素甲醚等4种蒽醌类化合物含量的测定方法,为完善该制剂的质量标准奠定基础。
1 材料与方法
1.1 仪器与试药 1525型HPLC仪,配2996型二极管阵列检测器(美国Waters公司),AL-204 电子天平(梅特勒-托利多),HH-2 型恒温水浴锅(常州国华电器有限公司)。
大黄酸(含量:99.8%,批号:0757-200005)、大黄素(含量:99.8%,批号:110756-200110)、大黄酚(含量:99.8%,批号:110796-201319)、大黄素甲醚(含量:99.8%,批号:110758-201013)对照品购自中国食品药品检定研究院,均为供含量测定用。甲醇(色谱纯,山东禹王实业有限公司),硫酸、磷酸、丙酮均为分析纯,水为二次蒸馏水。牛黄清火丸(规格:3g,批号:20130808,内蒙古天奇中蒙制药股份有限公司产品)购自鞍山巨力大药房。
1.2 方法
1.2.1 色谱条件与系统适用性试验:色谱柱:Kromasil C18(5 μm,150 mm×4.6 mm);流动相:甲醇-0.5%磷酸水溶液(75:25,V/V);流速:1.0 mL/min;检测波长:230 nm;柱温:30 ℃;进样量:10 μL。理论塔板数按照大黄酸、大黄素、大黄酚、大黄素甲醚算分别是9035、8804、6946、4073,对照品溶液、供试品溶液及阴性对照溶液色谱图,见图1。

图1 牛黄清火丸中大黄酸、大黄素、大黄酚、大黄素甲醚的高效液相色谱图A:对照品;B:供试品;C:阴性对照1.大黄酸;2.大黄素;3.大黄酚;4.大黄素甲醚Fig.1 HPLC chromatograms of rhein, emodin, chrysophanol andphyscion in Niuhuang Qinghuo PillsA:reference substance; B:sample; C:sample without rheum officinale1.rhein; 2.emodin; 3.chrysophanol; 4.physcion
1.2.2 溶液的制备:①对照品溶液的制备:分别精密称取大黄酸(3.0 mg)、大黄素(1.9 mg)、大黄酚(2.6 mg)、大黄素甲醚(2.3 mg)分置10 mL棕色量瓶中,乙醇溶解并稀释至刻度,得大黄酸(300 mg/L)、大黄素(190 mg/L)、大黄酚(260 mg/L)、大黄素甲醚(230 mg/L)对照品储备液。
分别移取4种对照品储备液各0.1 mL置同一10 mL棕色量瓶中,乙醇稀释至刻度,制得大黄酸(3.0 mg/L)、大黄素(1.9 mg/L)、大黄酚(2.6 mg/L)、大黄素甲醚(2.3 mg/L)混合对照品溶液。
②供试品溶液的制备:精确称取经剪刀剪碎的样品约1.0 g,置于装有冷凝器的100 mL圆底烧瓶中,加入20%的硫酸水溶液20 mL,沸水浴下回流水解30 min,将圆底烧瓶中的样品倒入布氏漏斗中,抽滤除去水解液,滤渣用蒸馏水清洗,滤纸和滤渣在80 ℃下烘干后,以丙酮为溶剂进行索氏提取,60 ℃水浴提取30 min,提取液转移至100 mL量瓶中,稀释至刻度为供试品溶液。
③阴性对照溶液的制备:按照处方(《中华人民共和国卫生部药品标准(中药成方制剂)》第七册WS3-B-1292-93)比例称取除大黄以外的其余八味药,按牛黄清火丸的制备工艺制备缺大黄的阴性样品,按照步骤②方法,制得阴性对照溶液。
1.2.3 标准曲线的绘制:分别取“1.2.2①”条下的4种对照品储备液0.05、0.1、0.5、1.0、5.0 mL置10 mL棕色量瓶中,乙醇稀释至刻度,摇匀,得系列标准溶液。按“1.2.1”条色谱条件进样分析,以浓度C(mg/L)为横坐标,峰面积A为纵坐标,进行线性回归。
1.2.4 精密度试验:取“1.2.2①”条下的混合对照品溶液,按“1.2.1”项下色谱条件重复进样测定6次,记录峰面积,计算含量及相对标准偏差。
1.2.5 重复性试验:按照步骤1.2.2②,制备供试品溶液6份,按“1.2.1”项下色谱条件进行测定,记录峰面积,计算含量及相对标准偏差。
1.2.6 稳定性试验:取“1.2.2②”条下的供试品溶液,按“1.2.1”项下色谱条件,分别于0、2、4、8、24 h进行测定,记录峰面积,计算含量及相对标准偏差。
1.2.7 加标回收率试验:精确称取样品约0.5 g 3份,分别加入不同质量的4种对照品,按照步骤1.2.2②制备供试液,HPLC测定含量,计算各对照品的加标回收率。
1.2.8 样品含量测定:取“1.2.2②”条下制备的供试品溶液,HPLC进行测定,检测3次,利用外标法测定其含量。
2 结果
2.1 标准曲线 以浓度C(mg/L)为横坐标,峰面积A为纵坐标得回归方程分别为:大黄酸A=5.813×104C+5.061×103(r=0.9983),大黄素 A=4.107×104C+1.997×103(r=0.9990),大黄酚A=3.617×104C+2.122×103(r=0.9990),大黄素甲醚A=5.499×104C+5.966×103(r=0.9950)。结果表明,大黄酸、大黄素、大黄酚和大黄素甲醚质量浓度分别在1.50~150、0.950~95.0、1.30~130和1.15~115 mg/L范围内与峰面积呈良好的线形关系。
2.2 精密度试验 大黄酸、大黄素、大黄酚、大黄素甲醚的RSD分别为1.17%、0.97%、1.04%和1.08%(n=6),结果表明仪器精密度良好。
2.3 重复性试验 大黄酸、大黄素、大黄酚、大黄素甲醚的RSD分别为0.86%、1.00%、0.45%和1.06%(n=6),说明本方法重复性良好。
2.4 稳定性试验 大黄酸、大黄素、大黄酚、大黄素甲醚的RSD分别为1.15%、0.80%、1.04%和0.92%(n=5),结果表明供试品溶液在24 h内稳定。
2.5 加标回收率试验 各对照品的加标回收率结果见表1。
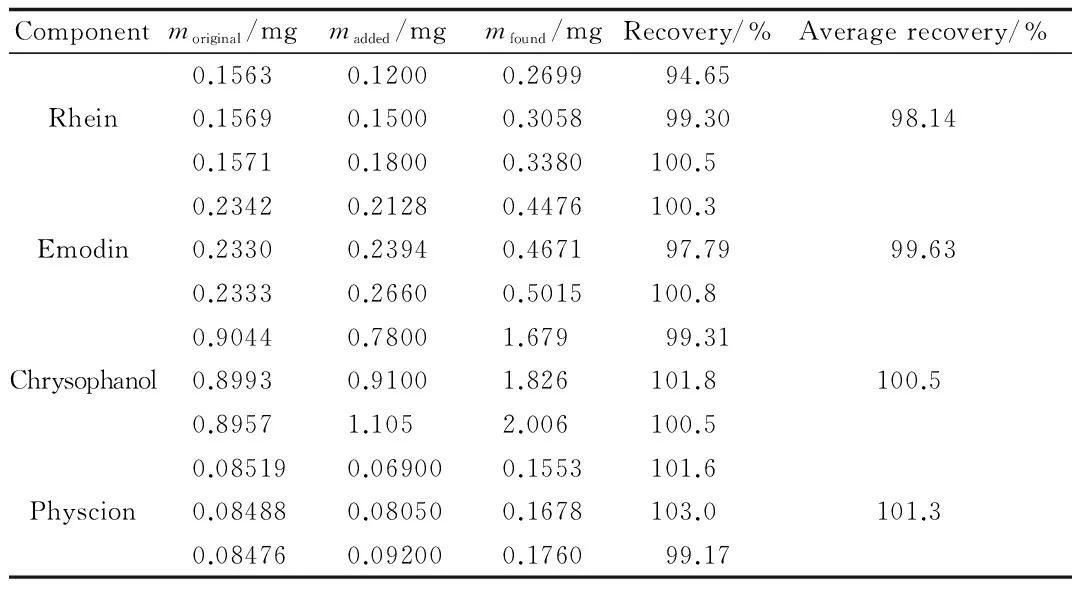
表1 加标回收率实验Tab.1 Results of recovery
2.6 样品含量测定 外标法测定样品含量结果见表2。

表2 样品含量测定结果(n=3)Tab.2 Results of content determination of sample
3 讨论
本文采用20%硫酸对样品进行水解,使结合态的蒽醌类化合物转化为游离态,增加样品测定的准确性。比较了二氯甲烷、无水乙醇、甲醇、丙酮、 乙醚等不同的索氏提取溶剂, 发现乙醇、甲醇沸点较高,回流需要较高的温度,二氯甲烷、乙醚提取时间长,并且提取不完全,在所考察溶剂中,丙酮的提取效果最为理想,并且在加标回收率试验中证明丙酮提取比较完全,因此选用其作为提取溶剂,并对提取温度和提取时间进行考查,最后确定60℃水浴提取30 min。
HPLC进行含量测定,采用甲醇-0.5%磷酸水溶液体系作为流动相,磷酸作为酸剂调节色谱峰的峰形,提高分离度,甲醇作为有机改性剂。甲醇的比例太高,大黄酸峰与杂质峰分离效果不好,若甲醇比例太低,延长分析时间,当甲醇:0.5%磷酸水溶液=75:25时,4种蒽醌类化合物都能得到较好的分离,最后选择其为流动相。
[1] 中华人民共和国卫生部药典委员会.中华人民共和国卫生部药品标准-中药成方制剂(第七册)[S].北京:中华人名共和国卫生部,1993:25.
[2] 苟奎斌,孙丽华,娄卫宁,等.大黄中4种蒽醌类化合物抑幽门螺杆菌效果比较[J].中国药学杂志,1997,32(5):24-26.
[3] 郑言博,马卓.蒽醌类化合物抗菌与抗肿瘤活性的研究进展[J].湖北中医杂志,2012,34(2):74-76.
[4] 梁荣感,罗伟生, 李利亚,等.大黄蒽醌类化合物体外抗流感病毒作用的研究[J].华夏医学,2006,19(3):396-398.
[5] 许乾丽,茅向军,宋晓宁,等.HPLC法同时测定六味安消胶囊中芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚的含量[J].药物分析杂志,2010,30(10):1841-1844.
[6] 靳丹虹,梁芳慧,李敬筠,等.微波辅助提取-HPLC测定决明子中的5种蒽醌类化合物[J].分子科学学报,2007,23(6):429-433.[7] 李鑫楠,黄毅岚,张丹,等.RP-HPLC测定熊胆降热丸中的5种大黄蒽醌类化合物[J].华西药学杂志,2007,22(6):697-698.
[8] 鲍蕾蕾,吴岩斌,秦路平,等.RP-HPLC法同时测定巴戟天中4种蒽醌类化合物的含量[J].中国药房,2010,21(15):1385-1387.[9] 高晓燕,卢建秋.HPLC-DAD法同时测定大黄中7个蒽醌类化合物的含量[J].药物分析杂志,2010,30(9):1636-1641.
[10] 李磊,袁波,李发美.HPLC法测定复方大青叶注射液中大黄素和大黄素甲醚的含量[J].沈阳药科大学学报,2004,24(1):38-40.
(编校:王俨俨)
Determination of anthraquinones in Niuhuang Qinghuo Pills by HPLC
DIAO Quan-ping, GUO Hua, LV Lin-lin, LI Tie-chun, HOU Dong-yanΔ
(College of Chemistry and Life Science, Anshan Normal University, Anshan 114007, China)
ObjectiveTo establish the method of determination for rhein, emodin, chrysophanol and physcion in Niuhuang Qinghuo Pills. MethodsSample was hydrolyzed by 20% H2SO4, and anthraquinones were extracted by soxhlet extraction with acetone as solvent, and were determined by HPLC with Kromasil C18as column, methanol-0.5%H3PO4(75:25,V/V)as mobile phase at the flow of 1.0 mL/min, 230 nm as the detection wavelength. ResultsThe linear relationship of rhein, emodin, chrysophanol and physcion were 1.50-150, 0.950-95.0, 1.30-130 and 1.15-115 mg/L. The anthraquinones were seperated completely, the recovery of 4 anthraquinones were 98.14%-101.3%. ConclusionThis method is simple, accurate, steady, and could be used for the quality control of Niuhuang Qinghuo Pills.
Niuhuang Qinghuo Pills; anthraquinones; HPLC
刁全平,男,硕士,副教授,研究方向:天然产物及药物分析,E-mail:qpdiao@163.com;侯冬岩,通信作者,男,硕士,教授,研究方向:天然产物有效成分分析,E-mail:houdyas@163.com。
R 917
A
1005-1678(2015)12-0177-03
