正丙醇水溶液准弹性中子散射光谱的分子动力学模拟
2012-12-11赵东霞
张 霞 张 强,* 赵东霞
(1渤海大学化学化工与食品安全学院,辽宁锦州121000;2辽宁师范大学化学化工学院,辽宁大连116029)
正丙醇水溶液准弹性中子散射光谱的分子动力学模拟
张 霞1张 强1,*赵东霞2
(1渤海大学化学化工与食品安全学院,辽宁锦州121000;2辽宁师范大学化学化工学院,辽宁大连116029)
准弹性中子散射(QENS)光谱是获取溶液分子动力学性质的重要方法,但光谱解析模型的有效性和去耦合近似的合理性仍存在争议.本文利用分子动力学模拟方法获取纯水和正丙醇水溶液中羟基氢原子的自相关中间散射函数FS(Q,t)和去耦合近似函数FP(Q,t),以及相关性质来评价它们的合理性.结果表明,在低动量转移范围内平-转去耦合近似相对合理,水分子的平-转耦合贡献较小,混合溶液中水分子的平-转耦合项和转动项随动量转移Q值增大而增大,二者显现相互抵消趋势.对于混合溶液中的正丙醇羟基氢原子,由于FS(Q,t)和质心自相关中间散射函数FCM(Q,t)偏差较大,利用实验光谱直接拟合分子平动扩散系数是不合适的.三种平动模型获取的纯水和正丙醇水溶液分子平动扩散系数与实验结果一致,略高于Einstein均方位移方法所得结果.水分子在纯水和混合溶液中表现为跳跃转动,而不是连续转动.正丙醇分子存在转动各向异性,羟基氢原子沿羟基向量为跳跃转动,沿相对质心向量可近似为连续转动.模拟结果显示,高动量转移范围平-转耦合项贡献较大,直接拟合实验光谱获取分子转动扩散系数或弛豫时间是不合适的.鉴于低动量转移范围内转动和平转耦合贡献较小,以及二者的抵消作用,在此范围内获取水分子平动信息是现实可行的.
准弹性中子散射光谱;分子动力学模拟;非相干结构因子;自相关中间散射函数;跳跃扩散
1 引言
水溶液分子动力学行为一直是物理化学研究的重要内容,核磁共振(NMR)、12D红外(2D-IR)2,3和准弹性中子散射(QENS)光谱4,5作为重要实验手段,让我们对其有了深入和微观的认识.QENS具有较宽的时间和空间测量范围,能够同时得到分子平动和转动信息,5已被应用到有机分子水溶液、超冷水溶液和受限水溶液体系的水分子动力学行为研究.4,6,7但是,由于缺乏可靠准确的物理模型来解析实验数据,使得获取的分子动力学信息有限或不确定性较大,其获取的水分子转动弛豫时间与NMR和分子动力学(MD)模拟结果相差较大,1-4进而影响了QENS的应用.
QENS光谱通过分子中原子(通常为氢原子)的散射光谱来获取溶液中分子的动力学信息,实验测定的非相干结构因子S(Q,ω)(是动量转移长度Q和能量转移长度ω的函数,Q≈4πλ-1sinθ,λ和θ为衍射波长和衍射角度)可以通过自相关中间散射函数FS(Q, t)的傅里叶变换得到,FS(Q,t)通常被近似为原子伴随分子质心的平动项和相对质心的转动项的乘积,被称为去耦合近似函数FP(Q,t).光谱解析方程中一般基于两个基本假设:一个是平动和转动去耦合(decoupling),如氢原子沿v方向运动,可近似为沿vCM方向分子整体质心平动和绕质心的相对转动vR的乘积(见图1A),且二者不存在耦合(实际耦合程度与分子和溶液微观结构有关);另一个为氢原子转动需满足连续扩散模型.8,9QENS最初利用连续平动和连续转动模型获取分子平动和转动扩散系数或弛豫时间,5但近年来大量的实验和理论研究表明,水溶液中水分子采用跳跃扩散方式10(见图1B,氢原子通过氢键交换反应完成沿箭头方向快速跳跃转动,此期间三个水分子质心框架O*O1O2保持相对不变,而后经历了氢键壳层的交换,发生跳跃平动),跳跃扩散模型12a被引入平动项在一定程度上提高了光谱解读能力,被称之为标准模型.11,12根据模耦合理论(MCT),水分子在超冷水溶液中存在明显的笼效应,13据此Chen等14提出了笼弛豫模型(RCM),能够更好地拟合实验结果,通常在低动量转移Q范围和超冷水溶液体系较为适用.15,16Qvist等17利用不指定拟合函数形式的无模型方法(model free approach)拟合实验光谱数据,发现水分子存在局域和非局域两种平动方式,非局域跳跃是水分子的主要平动方式.分子模拟研究表明,平动和转动去耦合近似在低Q值时通常相对合理,而在较大Q值时误差较大,中子散射光谱不能提供有效的分子转动信息.17实验和分子模拟结果表明,跳跃转动是水分子的主要运动方式(见图1B),不同程度违背传统的各向同性Debye连续扩散模型,8更符合Laage和Hynes18提出的扩展跳跃模型(EJM);Chen等14认为水分子转动贡献较小,可能与平-转耦合项相互抵消.评价转动和平-转耦合项的作用,探讨QENS光谱拟合方程的适用性,以及如何引入跳跃转动方式是当前QENS光谱解析的主要问题.目前,QENS光谱主要研究水分子的动力学性质,对非水溶液和多元混合溶液的动力学性质研究较少,分子动力学模拟可以直接计算中间散射函数和其分解项,是模型评价和实验数据解析非常有效的理论方法,为提高解析能力提供理论指导.
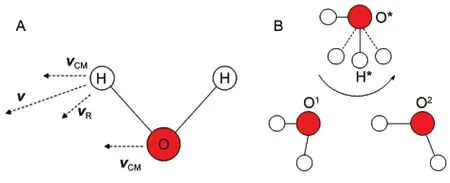
图1 分子和原子运动模式Fig.1 Modes of the molecular and atomic motions(A)The moving vector vof one hydrogen atom of water molecule and its decomposed components(the translation of center-of-mass vCMand the rotation around the center-of-mass vR);(B)jump translation and rotation(a fast jump rotation of hydrogen H*during a hydrogen bond exchange reaction from O1to O2and a fast translational jump following it18)
醇-水混合溶液通常作为实验和理论研究模型化体系,用以研究溶质疏水水合和各向异性溶液性质,溶液结构和混合热力学等热点问题.19,20体系的动力学性质越来越受到关注,中子散射、介电光谱和分子模拟在解释微观结构和分子动力学性质方面作用日益显现.15,21,22本文以摩尔分数(xp)为0.167的正丙醇水溶液为研究对象,正丙醇和水分子具有不同的转动半径和分子极性,符合本文研究的模型要求,利用分子动力学模拟轨迹得到水和正丙醇羟基氢原子的自相关中间散射函数FS(Q,t)、去耦合近似函数FP(Q,t)、平动和转动贡献项,以及相关的平动和转动参数,对比和评价光谱拟合方程的有效性和适用性,分析分子运动规律,探讨溶质对溶剂水分子光谱的影响.浓度依赖递变性质不是本文重点,选用该混合比旨在分析混合的影响和做对照分析.15
2 准弹性中子散射与分子动力学模拟细节
2.1 准弹性中子散射
自相关中间散射函数FS(Q,t)可以通过分子动力学模拟轨迹直接计算得到,通过傅里叶变换转变为与实验对应的准弹性中子散射S(Q,ω),即非相干动力学结构因子(ISF),5,12,14,15
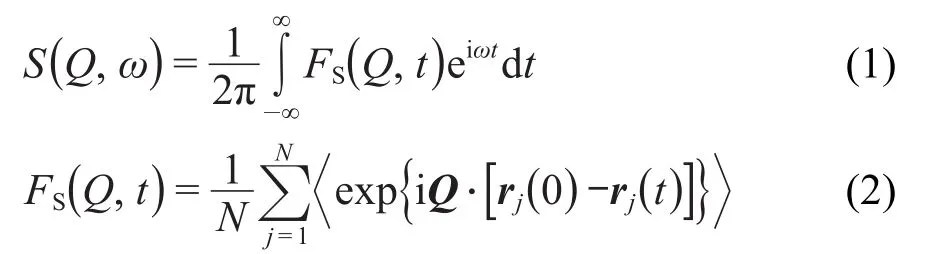
上式中,N是体系水分子的氢原子个数,rj(t)是第j个氢原子的位置向量,S(Q,ω)是动量转移Q和能量转移ω的函数,相应波矢Q=2π/L(n,m,l),L为模拟盒长,n、m和l为整数.为了获取氢原子的平动和转动信息,通常将其分解为振动、平动和转动三个部分,同时加入平转耦合项STR(Q,ω),
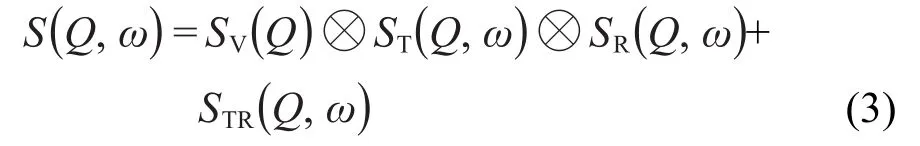
SV(Q)为非频率依赖性振动项,称之为Debye-Waller为振动均方移位,影响S(Q,ω)强度,由于分子模拟中使用刚性化学键近似,因而该项近似为1,后文中忽略了该项.ST(Q,ω)和SR(Q,ω)为非相干散射函数的平动和转动项.为了使理论和实验数据具有合理对应关系,将氢原子的位置向量分解为质心平动和绕质心相对转动部分,r=rCM+rd,rd为氢原子相对分子质心rCM的相对坐标,由于采用刚性分子构型,rd为氢原子绕质心转动半径,其取向rd=rd/rd.分解后的FS(Q,t)用平-转去耦合中间散射函数FP(Q,t)表示,12忽略平-转耦合项,

分解后的平动和转动部分分别与分子的平动和转动扩散系数相关联,12-14
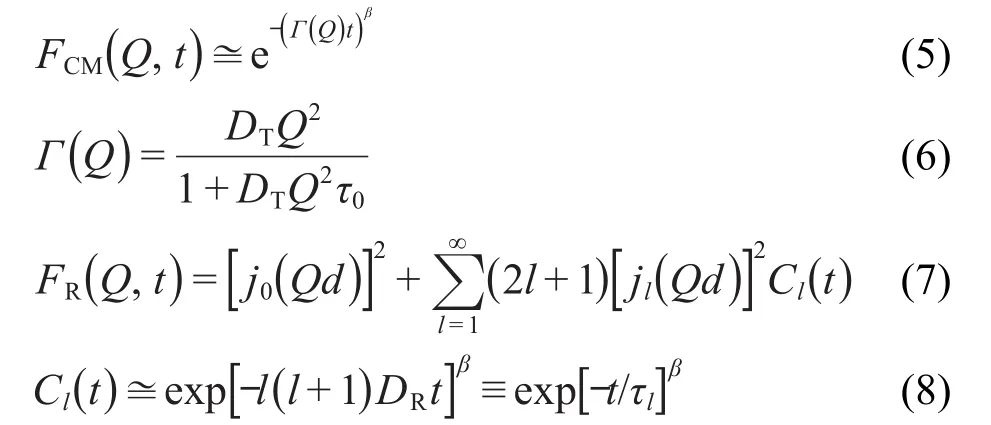
这里,Г(Q)为洛伦兹线宽,jl为第l阶球贝塞尔函数.方程(6)为平动跳跃模型,如果忽略分子平动和转动的各向异性,则e指数展宽因子,β值为1,此时FP(Q, t)被称之为标准模型.12-14DT、DR和τ0为平动扩散系数、各项同性转动扩散系数和跳跃弛豫时间.为向量rd重取向相关函数,Pl为勒让德多项式的第l项,τl为第l阶展开式的转动弛豫时间,实验拟合中可通过近似式τl=[l(l+1)· DR]-1获取DR.
FCM(Q,t)通常利用跳跃扩散模型来描述,从而获取DT和τ0,并通过跳跃长度L相关联,L=(6DTτ0)-1.如果考虑平动各向异性,则有效洛伦兹线宽在笼弛豫模型下为计算平动扩散系数.由于加入了额外展宽参数,提高了拟合准确度,但是很难转化为解析形式的S(Q,ω).
Qvist等17利用无模型方法拟合实验光谱,发现S(Q,ω)存在两个弛豫过程,FS(Q,t)符合双e指数函数规律,
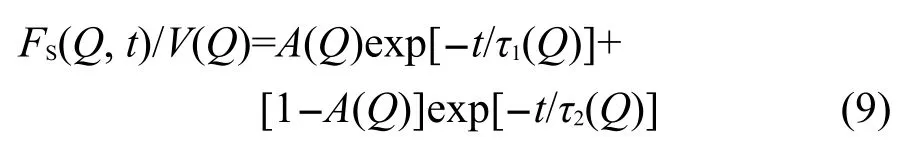
通过拟合方程(9)可得到参数τ1(Q)和τ2(Q),二者与物理微观弛豫过程相对应,水分子平动可分为微观受限结构域间的跳跃(弛豫时间为τJ)和结构域内浮动(弛豫时间为τL),其关系为τJ=τ1,1/τL=1/τ2-1/τ1,τJ对应慢弛豫控制过程,τL对应快过程,A(Q)为两种弛豫模式的强度因子.方程拟合得到的参数有明确的微观物理意义,平动扩散系数通过方程Γ(Q)=1/τJ(Q)= DLJQ2/(1+(Qd)2/τ0)在低Q值范围内拟合得到,DLJ和d分别为分子平动扩散系数和跳跃长度,他们在工作方程中忽略了转动贡献,认为转动项和平-转耦合项相互抵消,A(Q)的物理意义相当于标准模型中的[j0(Qd)]2.
标准模型下,非相干结构因子S(Q,ω)由两个洛伦兹函数加和组成,其表达为12
方程右边第一项为平动贡献项ST(Q,ω),第二项为转动贡献SR(Q,ω),如果考虑大角度分子跳跃转动,则用转动弛豫时间的倒数1/τl代替近似项连续转动扩散参数[l(l+1)DR].实验光谱通过拟合该方程得到分子平动和转动扩散系数或弛豫时间.
2.2 分子动力学模拟细节
分子模拟采用普遍认可的SPC/E水分子力场,23正丙醇分子采用OPLS-UA力场几何构型,24原子位点电荷参数参照H1模型,25该模型能够较好地模拟甲醇水混合溶液的动力学性质,26非键相互作用参数见表1.由于氢原子散射函数,以及它的平动、转动项都源于同一模拟轨迹,具有自一致性,因而忽略力场参数对结果影响.中心盒子中的500个分子按照正丙醇摩尔分数xP=0.167随机填充,模拟中保持水分子刚性结构,正丙醇分子只约束键长和键角为平衡参数值,其他自由度未限制.采用Maxwell-Boltzmann分布方法获得体系原子或分子初始速度,利用Berendsen热浴方法27控制体系温度,控温弛豫时间为0.1 ps.体系中粒子的运动轨迹采用蛙跳方法28积分得到,时间步长为2 fs.模拟体系密度均与实验一致.非键范德华相互作用的截断半径设定为1.0 nm,静电相互作用利用Ewald求和方法29得到,最近镜像方法使体系保持连续性.对于每一个体系,均采用NVT系综下1 ns平衡模拟,利用后续NVE系综下2 ns平衡模拟轨迹获取所需的物理量.纯水体系的相应模拟结果作为对照.分子动力学模拟在Tinker软件包30下完成.

表1 正丙醇分子力场参数和几何构型Table 1 Force field parameters and molecular geometries of 1-propanol
3 结果与讨论
我们利用分子动力学模拟轨迹分别计算水分子和正丙醇分子氢原子的自相关中间散射函数FS(Q,t),同时利用分解方法计算它们相应的平动和转动的散射函数FCM(Q,t)和FR(Q,t),实验测定的Q值范围为3.76-25.04 nm-1,模拟中我们测定Q值的范围为2.2-23.1 nm-1.SPC/E采用刚性水分子结构,其氢原子平均转动半径为0.0964 nm,实验值为0.098 nm,11正丙醇氢原子的平均转动半径为0.215 nm.
通常利用准弹性中子散射函数方程(10)来获取溶液中氢原子的平动和转动信息,从而预测所属分子的活动性.应用该方法的合理性要基于两种基本假设:一个是平动和转动去耦合,另一个为氢原子转动应符合连续扩散模型.之外,转动和平-转耦合项是否可以定量相互抵消,进而在标准模型下忽略转动贡献,也是我们要探讨的问题.
首先我们对纯水氢原子(W0)、xP=0.167混合溶液中水氢原子(W1)和丙醇羟基氢原子(P1)的FS(Q, t)、FCM(Q,t)、FR(Q,t)和FP(Q,t)进行对比分析,选取三个特定动量转移长度Q=7 nm-1、10 nm-1和14 nm-1,分别列于图2(A-C).结果表明,W0、W1和P1的平-转耦合程度[FS(Q,t)-FP(Q,t)]和转动贡献[FCM(Q, t)-FP(Q,t)]都随Q和t增大而增大,其中W0差异最小,P1最大,同一Q值时三种氢原子中间散射函数也具有相同大小趋势.虽然纯水的质心平动相关消减较快,但是由于其转动半径较小,FR(Q,t)消减较慢(理论极小值为[j0(Qd)]2),导致FSW0(Q,t)、FPW0(Q,t)和和合近似.W0的随Q值增大有相互接近趋势,W1也有类似情况,而和恰相反,W0和W1的转动贡献项不断增大,同时,平转耦合项也朝相反方向增大,二者定量相互抵消趋势明显,因此对于W0和W1而言,利用FS(Q,t)代替FCM(Q,t)来拟合平动扩散系数也存在数值合理性问题.10,18但对含有较大疏水基团和转动半径的P1而言,利用分解方法获取光谱信息显然缺乏依据,以此得到的结果无实际意义.
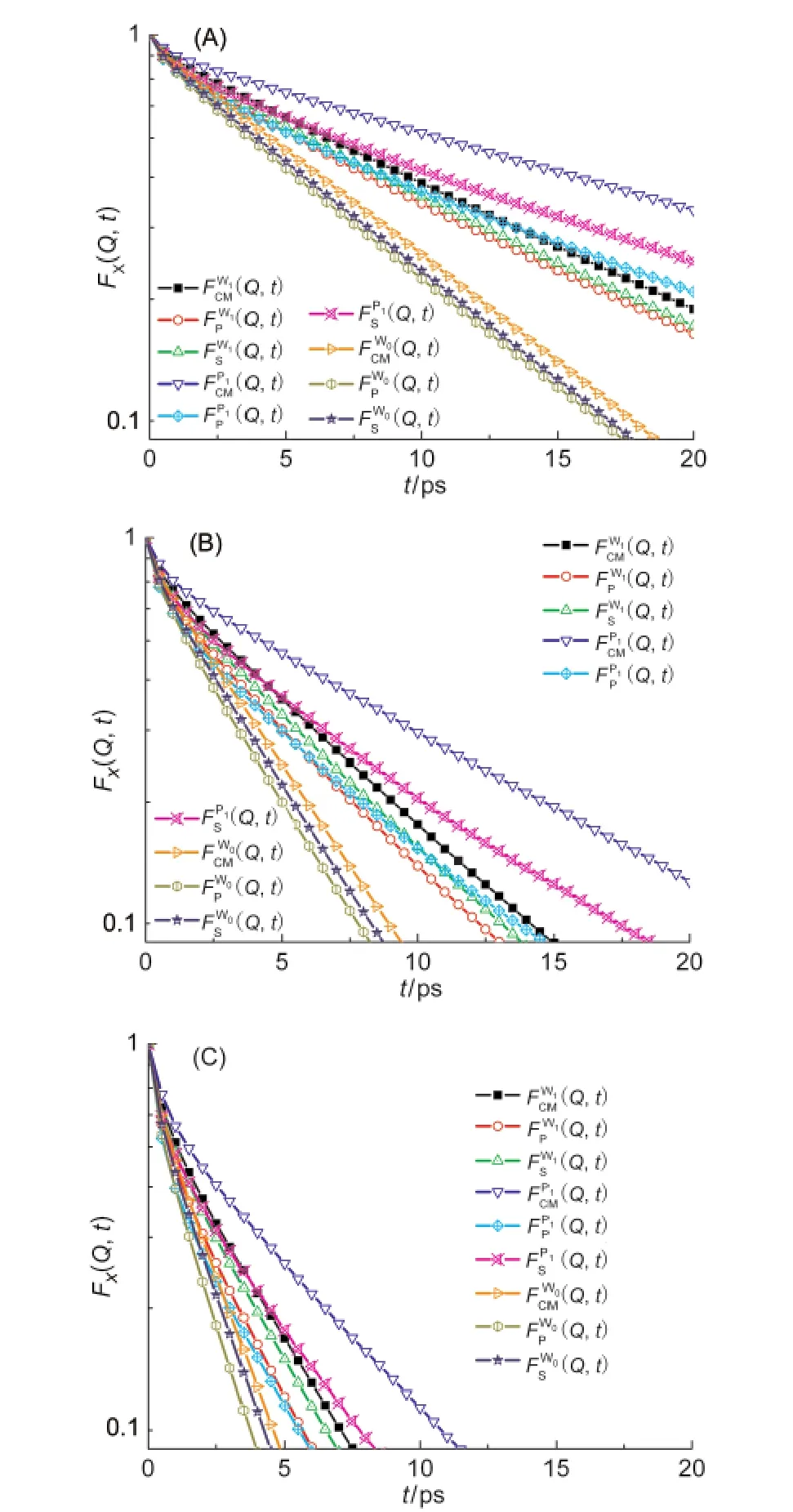
图2 Q值为7 nm-1(A)、10 nm-1(B)和14 nm-1(C)的自相关中间散射函数FS(Q,t)、FCM(Q,t)、FR(Q,t)和FP(Q,t)Fig.2 Self-intermediate scattering functions FS(Q,t), FCM(Q,t),FR(Q,t),and FP(Q,t)at Q=7 nm-1(A), 10 nm-1(B),and 14 nm-1(C)The superscripts W0and W1denote the water hydrogen in pure water and 1-propanol/water mixture,respectively,and P1means the hydroxyl hydrogen of 1-propanol in the mixture.
由于在低Q值范围内散射光谱主要由平动控制,所以在此范围内利用实验光谱和方程(10)可以拟合得到洛伦兹线宽Г(Q),再通过平动跳跃模型方程(6)获取测定原子所属分子的平动扩散系数;也可以利用分子动力学模拟轨迹直接计算FCM(Q,t),根据选定的平动模型方程(如跳跃扩散模型、笼弛豫模型、无模型方法等)拟合FCM(Q,t),获取相应的Г(Q)或最终得到分子平动扩散系数.FCM(Q,t)的拟合范围为t>1 ps,去掉短时间分子摆动弛豫过程,笼弛豫模型的扩散系数在4.84 nm-2<Q2<50 nm-2范围内拟合得到.图3中给出了不同模型下计算得到的Г(Q)或在2.2 nm-1<Q<10 nm-1时,Г(Q)与Q2呈较好的线性关系,随着Q值增大,水分子的Г(Q)向某一极限值收敛,这是水分子跳跃平动的结果,4而对于P1三种模型下均未出现明显收敛弯曲,说明丙醇分子没有明显的跳跃平动行为.高Q值区域,展宽e指数获取的平动弛豫时间相对较短,Qvist等17认为展宽e指数虽然具有良好的数值拟合能力和对超冷低温体系有很好的描述,但是仍是对单一α弛豫微观过程的描述,而且弛豫时间和展宽指数具有一定依赖性.2.2 nm-1<Q<10 nm-1范围内三种平动模型下的Г(Q)值没有明显区别,三种模型下的纯水扩散系数和DLJ分别为2.53×10-5、2.55×10-5和2.50×10-5cm2·s-1,质心均方位移随时间变化斜率为2.44×10-5cm2·s-2,略高于Einstein均方位移方法,在二甲基亚砜水溶液中也存在相同的趋势,12相应的实验结果在2.2×10-5-2.6×10-5cm2·s-2之间,Laage11用标准模型得到水的扩散系数为2.3×10-5cm2·s-2(293 K),Nakada等15利用实验光谱和笼弛豫模型获得的扩散系数为2.32×10-5cm2·s-2(298 K).对于混合溶液中水分子和丙醇分子平动扩散系数,三种平动模型与Einstein方法结果15,31符合较好(表2),与实验结果的偏差主要来源于分子力场方法,以及实验结果的不确定性.图4列举了利用笼弛豫模型得到的展宽因子变化曲线,β随Q值增大而减小,在混合溶液中变化幅度增大,体现了混合体系分子环境各向异性的突出特点,与多肽水溶液体系的递变规律具有一致性,16与Nakada等15结果存在差异,其原因主要归结为实验光谱拟合时,其转动部分采用了连续扩散模型近似,以及固定转动弛豫时间和e指数展宽因子累计形成.以上分析表明,对于丙醇水溶液体系,三种平动模型都能较好地获取水和丙醇分子平动信息,无模型方法和跳跃扩散模型能够合理描述洛伦兹线宽极限趋势.
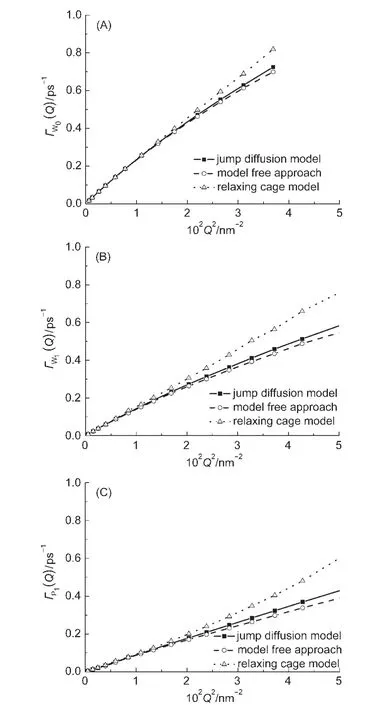
图3 三种平动模型下的洛伦兹线宽Г(Q)Fig.3 Lorentzian linewidth Г(Q)of three translation models(A)water in pure liquid water,(B)water in mixture, (C)1-propanol in the mixture

图4 e指数展宽因子βFig.4 Stretch exponential factor β
转动项部分一直是解析QENS光谱的关键问题,在高Q值范围内转动贡献增大,光谱强度由Г(Q)+[l(l+1)DR]两项决定.平动和转动项相互依存.我们根据相关函数计算了W0、W1和P1的质心向量(MH)和羟基向量(OH)的转动重取向弛豫时间,结果列于表2中.我们发现水分子的羟基向量和质心向量转动弛豫时间基本一致, W0和W1的和均接近于2,而连续转动扩散模型下其理论值为3,从而说明水分子不符合连续扩散行为,更倾向于跳跃转动方式.11混合溶液中水分子的平动扩散系数降低,转动弛豫时间增加,主要因为丙醇分子的空间位阻和疏水作用降低了水分子的平动和转动有效体积,水分子在溶质周围热力学熵降低.丙醇分子的和分别为1.97和2.59.质心向量和羟基向量重取向转动明显不同,羟基转动呈现跳跃特征,质心转动接近连续扩散行为,同时具有疏水和亲水基团的丙醇分子转动存在明显的各向异性.所以,QENS的转动部分采用连续扩散模型对于本文研究体系显然是不合适的,如果采用方程(10)拟合平动和转动参数,在高Q范围内很难得到有意义的结果.通过图2的比较和前面的分析,纯水体系的转动贡献在常态下相对较小,在低Q值范围利用S(Q,ω)拟合平动扩散系数相对合理,可以忽略转动部分,而对于丙醇水溶液而言,显然缺乏依据.高Q值范围内W0、W1和P1的平-转耦合项贡献都较大,不能简单地直接利用实验S(Q,ω)代替SP(Q,ω).
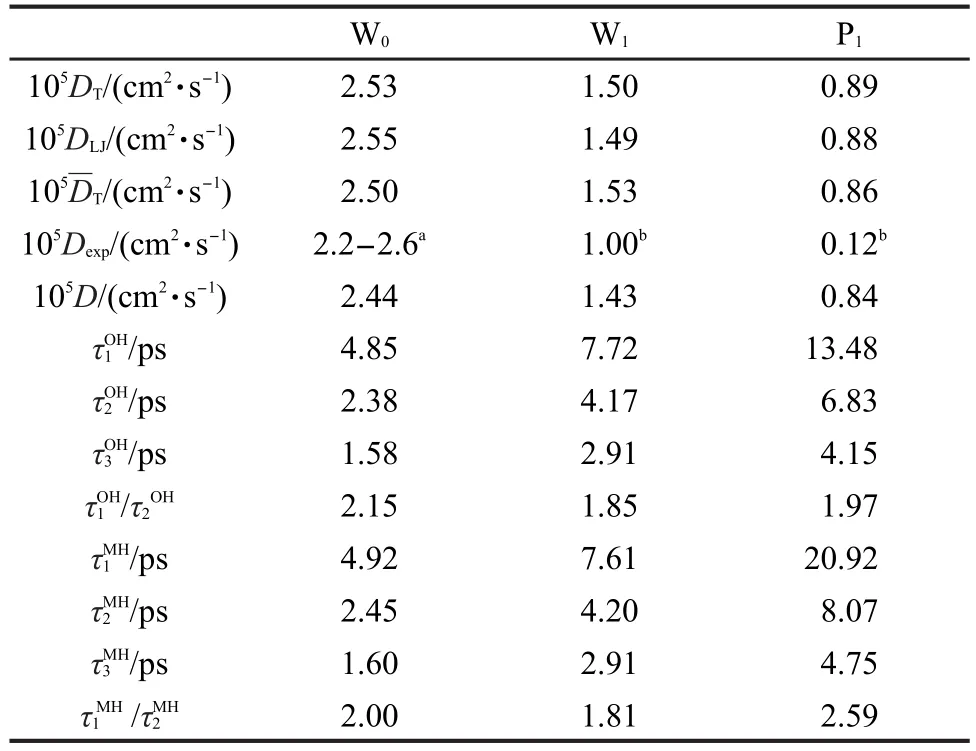
表2 平动扩散系数和转动弛豫时间Table 2 Translation diffuse constants and rotation relaxation time

图5 非相干结构因子Fig.5 Incoherent structure factors(A)the incoherent structure factors SS(Q,ω)of three types of hydrogen;The incoherent structure factors SS(Q,ω)、SP(Q,ω)、ST(Q,ω),and SR(Q,ω) for(B)the water hydrogen in the pure water,(C)the water hydrogen in the mixture,and(D)the hydroxyl hydrogen of 1-propanol in the mixure
分子模拟得到的自相关中间散射函数FS(Q,t)和FP(Q,t),经过傅里叶变换得到相应的非相干结构因子SS(Q,ω)和SP(Q,ω),并根据方程(10)和相应参数得到ST(Q,ω)和SR(Q,ω),图5中列举了Q=10 nm-1时,三类氢原子相应的非相干结构因子(非相干结构因子与光谱的分辨函数乘积相当于实验光谱).通过比较图5A和表1的结果可知,分子活动性降低,光谱分布宽度变小,混合溶液中的氢原子活动性明显降低,丙醇分子活动性低于水分子.水分子的平-转去耦合近似在Q=10 nm-1时相对合理,W0和W1的SS(Q,ω)和SP(Q,ω)差别较小,而丙醇分子显然偏差较大;水分子的平动项在此动量转移值时是主要贡献项(图5B和5C),具有较大转动半径的丙醇分子则由转动项控制(图5D).通过对非相干结构因子的直观分析表明,光谱解析方程的近似形式和模型的选取对分子动力学信息的获取有重要影响.
4 结论
QENS光谱是获取溶液中分子动力学信息的重要手段,但由于解析光谱困难使其应用受到限制.本文通过对纯水和丙醇水溶液中水和丙醇羟基氢原子的FS(Q,t)、FP(Q,t)、FCM(Q,t)和FR(Q,t)的对比分析表明,水分子的转动贡献较小,在2.2 nm-1<Q<10 nm-1范围内平转去耦合近似相对合理,混合效应使水分子的平-转耦合略有增强,随着Q值增大,转动和平转耦合项相互抵消趋势显现.对于丙醇分子,去耦合近似在整个Q值范围内均不合理,转动贡献较大,不能简单将实验光谱近似为平动光谱.我们利用三种平动模型拟合计算了不同Q值下的洛伦兹线宽和平动扩散系数,通过比较发现,三种模型都能够很好地获取纯水体系水分子的平动扩散系数,而标准模型和无模型方法对混合溶液中的分子平动描述也相对合理,得到的平动扩散系数与Einstein方法一致,而笼驰豫模型获取的结果略高于其他方法.水分子的转动表现为跳跃行为,丙醇分子的疏水作用使水分子活动性降低,但未改变水分子跳跃转动特性,丙醇分子表现为各向异性转动性质,羟基为跳跃转动,绕分子质心转动可近似为连续转动.由于在高Q值时平-转耦合项的存在和贡献增大,利用实验光谱很难获取合理的分子平动和转动信息,利用低Q范围获取水分子平动信息是现实合理的.
(1) Ropp,J.;Lawrence,C.;Farrar,T.C.;Skinner,J.L.J.Am. Chem.Soc.2001,123,8047.
(2) Rezus,Y.L.A.;Bakker,H.J.J.Chem.Phys.2005,123,114502.
(3) Park,S.;Moilanen,D.E.;Fayer,M.D.J.Phys.Chem.B 2008, 112,5279.
(4) Cabral,J.T.;Luzar,A.;Teixeira,J.;Bellissent-Funel,M.C. J.Chem.Phys.2000,113,8736.
(5) Bée,M.Quasielastic Neutron Scattering;Adam Hilger:Bristol, 1988.
(6) Teixeira,J.;Luzar,A.;Longeville,S.J.Phys.:Condens.Matter 2006,18,S2353.
(7) Harpham,M.R.;Ladanyi,B.M.;Levinger,N.E.;Herwig,K. W.J.Chem.Phys.2004,121,7855.
(8) Debye,P.Polar Molecules;The Chemical Catalog Company: New York,1929.
(9) Sears,V.F.Can.J.Phys.1966,44,1299.
(10) Teixeira,J.;Bellissent-Funel,M.C.;Chen,S.H.;Dianoux,A.J. Phys.Rev.A 1985,31,1913.
(11) Laage,D.J.Phys.Chem.B 2009,113,2684.
(12) (a)Egelstaff,P.A.An Introduction to the Liquid State; Academic:London,1967. (b)Harpham,M.R.;Levinger,N.E.;Ladanyi,B.M.J.Phys. Chem.B 2008,112,283.
(13) Götze,W.;Sjögren,L.Rep.Prog.Phys.1992,55,241.
(14) Chen,S.H.;Liao,C.;Sciortino,F.;Gallo,P.;Tartaglia,P.Phys. Rev.E 1999,59,6708.
(15)Nakada,M.;Maruyama,K.;Yamamuro,O.;Misawa,M. J.Chem.Phys.2009,130,074503.
(16) Murarkaa,R.K.;Head-Gordon,T.J.Chem.Phys.2007,126, 215101.
(17) Qvist,J.;Schober,H.;Halle,B.J.Chem.Phys.2011,134, 144508.
(18) Laage,D.;Hynes,J.T.J.Phys.Chem.B 2008,112,14230.
(19) Dixit,S.;Crain,J.;Poon,W.C.K.;Finney,J.L.;Soper,A.K. Nature 2002,416,829.
(20) Sato,T.;Chiba,A.;Nozaki,R.J.Chem.Phys.2000,113,9748.
(21) Sato,T.J.Mol.Liq.2005,117,23.
(22) Roney,A.B.;Space,B.;Castner,E.W.;Napoleon,R.;Moore, P.B.J.Phys.Chem.B 2004,108,7389.
(23) Berendsen,H.J.C.;Grigera,J.R.;Straatsma,T.P.J.Phys. Chem.1987,91,6269.
(24) Jorgensen,W.L.;Maxwell,D.S.;Tirado-Rives,J.J.Am.Chem. Soc.1996,118,11225.
(25) Haughney,M.;Ferrario,M.;McDonald,I.R.J.Phys.Chem. 1987,91,4934.
(26) Chowdhuria,S.;Chandra,A.J.Chem.Phys.2005,123,234501.
(27) Berendsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.;Di Nola,A.;Hauk,J.R.J.Chem.Phys.1984,81,3684.
(28) Allen,M.P.;Tildesley,D.J.Computer Simulation of Liquids; Clarendon:Oxford,1987.
(29) Essmann,U.;Perera,L.;Berkowitz,M.L.;Darden,T.;Lee,H.; Pedersen,L.G.J.Chem.Phys.1995,103,8577.
(30) Ponder,J.W.;Richards,F.M.J.Comput.Chem.1987,8,1016.
(31) Hawlicka,E.;Grabowski,R.J.Phys.Chem.1992,96,1554.
December 20,2011;Revised:March 4,2012;Published on Web:March 7,2012.
Quasi-Elastic Neutron Scattering Spectroscopy of the 1-Propanol/Water Solution by Molecular Dynamics Simulations
ZHANG Xia1ZHANG Qiang1,*ZHAO Dong-Xia2
(1Institute of Chemistry,Chemical Engineering and Food Safety,Bohai University,Jinzhou 121000,Liaoning Province,P.R.China;2Institute of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
Quasi-elastic neutron scattering(QENS)spectroscopy as an important tool can be used to extract the molecular dynamic properties.However,the validity of the dynamical models and the decoupling approximation used in QENS spectral analysis is a topic of ongoing debate.In this paper,the self-intermediate scattering function FS(Q,t)and the decoupling approximation function FP(Q,t)of the hydroxyl hydrogen in pure water and in 1-propanol/water mixture,and certain dynamic properties predicted by three translation models,are derived from molecular dynamics simulations to assess their reasonability. The results suggest that the decoupling approximations for the water hydrogen in pure water and in mixture are reasonable at low momentum transfer Q.The contribution from the translation-rotation coupling term is small for the pure water.The coupling effect is strengthened for the water hydrogen when 1-propanol is added to the water.Under these conditions,the coupling and rotation terms both increase with the momentum transfer Q and largely cancel each other.For the hydroxyl hydrogen of 1-propanol in the mixture,the translational diffusion constant cannot be directly derived from the experimental spectrum,due to large deviation between FS(Q,t)and the center-of-mass translational function FCM(Q,t).The translational diffusion constants by the three translation models used in our current work are consistent with experimental results and a little higher than those predicted by the Einstein method.The jump rotation,as opposed to continuous rotation,is observed for the water molecule in both bulk water and mixture.For the 1-propanol molecule,rotations are anisotropic,being continuous along the axis from the hydroxyl hydrogen to the center-of-mass,and jumping along the hydroxyl bond vector.Simulations indicate that neither the rotational diffusion constant nor the relaxation time at high momentum transfer Q are adequately determined by the decoupling models,since the coupling effects become significant.Within the low momentum transfer range,the translation properties can be reasonably derived,due to the negligible contributions from the rotation and the coupling terms,as well as the canceling effect between them.
Quasi-elastic neutron scattering spectroscopy;Molecular dynamics simulation; Incoherent structure factor;Intermediate scattering function;Jump diffusion
10.3866/PKU.WHXB201203072
∗Corresponding author.Email:zhangqiang@bhu.edu.cn;Tel:+86-416-3400698.
The project was supported by the National Natural Science Foundation of China(20873055,21176029).
国家自然科学基金(20873055,21176029)资助项目
O643
