高效液相色谱法测定复方磺胺甲恶唑片中SMZ、TMP的含量
2011-10-30石向群郑由敏周凯凤孙利利
吕 璐,石向群,郑由敏,周凯凤,孙利利
(九江学院基础医学院,江西 九江 332000)
高效液相色谱法测定复方磺胺甲恶唑片中SMZ、TMP的含量
吕 璐,石向群,郑由敏,周凯凤,孙利利
(九江学院基础医学院,江西 九江 332000)
目的 建立测定复方磺胺甲恶唑片中磺胺甲恶唑(SMZ)和甲氧苄啶(TMP)含量的高效液相色谱(HPLC)法。方法 色谱柱C18(5 μm,4.6 mm ×150 mm),以0.03 mol/L磷酸(含0.2%三乙胺)-甲醇为流动相进行梯度洗脱,流速1.0 ml/min,检测波长为 240 nm,柱温 40℃,峰面积外标法。结果 SMZ 在 20~100 μg/ml有良好的线性,r=0.993 1;TMP 在 4~20 μg/ml有良好的线性,r=0.996 6。结论 此法简单、专属性强、重现性好、结果准确,可作为该片剂质量控制的参考方法。
高效液相色谱法;复方磺胺甲恶唑片;SMZ含量;TMP含量
复方磺胺甲恶唑片是临床常用的磺胺类抗菌消炎药,其主要成分为磺胺甲恶唑(SMZ)和甲氧苄啶(TMP)。《中国药典》(2000年版)采用等吸收双波长分光光度法对2组份分别进行测定。另有文献报道采用倍率减差双波长法、三波长法、吸收度线性线合法、双波长比值光谱法等方法测定其含量,但均存在操作或计算较为繁琐的缺点。现采用高效液相色谱法,对2组份SMZ、TMP同时进行测定。
1 实验部分
1.1 仪器
高效液相色谱仪(瓦里安Varian Prostar 230 SDM/325 UV);液相工作站(Varian Star 6.4);电子分析天平(赛多利斯Sartorius CP324S);高速离心机(中佳HC-2518);超声波清洗机(宁波新芝生物);无油隔膜真空泵(天津奥特赛恩斯)。
1.2 试药
对照品:磺胺甲恶唑、甲氧苄啶(中国药品生物鉴定所提供);复方磺胺甲恶唑片(SMZ 0.4 g、TMP 0.08 g,湖北华中药业有限公司生产);甲醇为色谱纯;其他试剂均为分析纯;水为超纯水。
1.3 步骤与条件
1.3.1 色谱条件 色谱柱是Varian C18(5 μm,4.6 mm×150 mm);流动相为甲醇:0.03 mol/L磷酸(含0.2%三乙胺);以甲醇比例20%~60%梯度洗脱;检测波长240 nm;流速1.0 ml/min;进样量 100 μl,定量环 20μl;柱温 40℃;样品分析时间 10 min。
1.3.2 对照品溶液的制备 精密称取经105℃干燥至恒重的对照品SMZ 12.5 mg、TMP 2.5 mg置于25 ml容量瓶中,加甲醇溶解并稀释至刻度。精密吸取对照品溶液0.4 ml、0.8 ml、1.2 ml、1.6 ml、2.0 ml分别置10 ml量瓶中,加甲醇使各容量瓶中溶液的体积为3 ml,加含20%甲醇的流动相定容至刻度,摇匀,配成标1、标2、标3、标4、标5混标溶液,以 15 000 r/min 离心 10 min,取上清液进样。
1.3.3 样品溶液的制备 取复方磺胺甲恶唑10片,精密称定,研细。精密称适量(约相当于SMZ 12.5 mg、TMP 2.5 mg)置25 ml量瓶中,加甲醇适量,经超声处理15 min后,加甲醇稀释至刻度。摇匀,滤过,弃去初滤液,精密量取续滤液1.6 ml,置10 ml量瓶中,参照2.2方法定容、离心、进样。
2 结果与讨论
2.1 保留时间试验
取标2和样品各一份,分别按100μl进样,按2.1色谱条件进行分析,结果标2的保留时间为SMZ 6.848 6 min、TMP 5.408 6min,样品的保留时间为SMZ6.8453min、TMP5.4089min,其HPLC色谱图见图1。
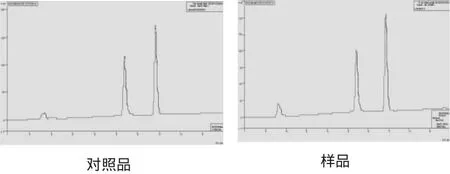
图1 对照品及样品HPLC色谱图
2.2 线性关系考察
分别取系列标准溶液100 μl,依次进样,按2.1色谱条件进行分析,分别测其峰面积,采用外标法,分别以SMZ和TMP峰面积Y为纵坐标,各组份的浓度X(mg/ml)为横坐标,进行线性回归,得回归方程,相关系数及线性范围分别为SMZ:Y=+1.650 0 e+008 x+6.636 0 e+005,r=0.993 1,线性范围为 20~100 mg/ml;TMP:Y=+5.890 2 e+008 x+2.168 5 e+006,r=0.996 6,线性范围为4~20 mg/ml。对照品溶液标准曲线见图2。
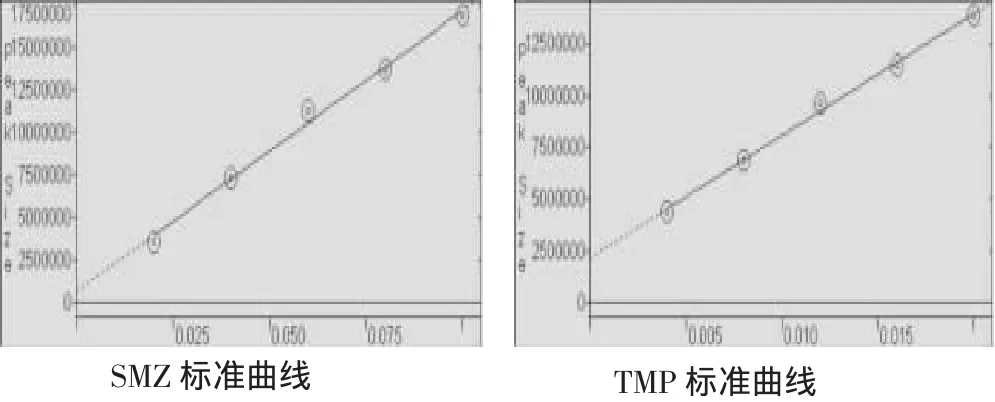
图2 对照品标准曲线
2.3 精密度试验
取标3对照品溶液100 μl,重复进样3次,按照2.1色谱条件进行分析,计算含量,结果见表1。

表1 精密度试验结果
2.4 样品含量测定
分别取3份样品溶液进样,按照2.1色谱条件进行分析,计算含量,结果见表2。
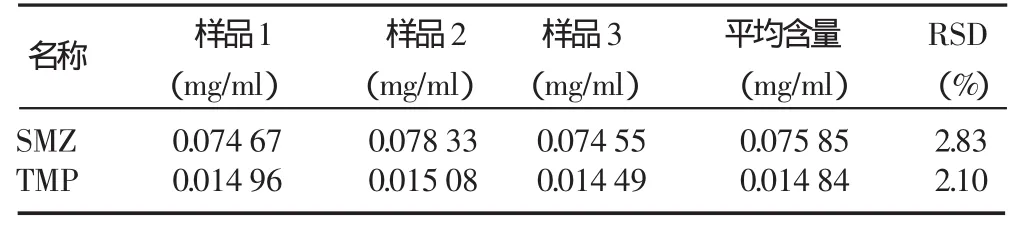
表2 样品试验结果
测得的SMZ和TMP含量分别是复方磺胺甲恶唑片标示量的94.81%和92.75%。
2.5 加标回收试验
在样品中加入一定量的对照品母液,配置成3份加标溶液,分别进样,按2.1色谱条件进行分析,做加标回收试验,计算回收率。结果见表3。
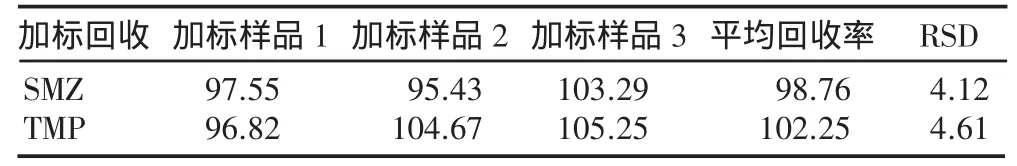
表3 加标回收试验结果(%)
2.6 讨论
2.6.1 检测波长的选择 复方磺胺甲恶唑片中SMZ、TMP的规格分别为4 mg/片、0.8 mg/片,含量相差较大,故本试验选择TMP紫外吸收度较大的波长(240 nm)为检测波长,以提高TMP测定的灵敏度和准确性。
2.6.2 洗脱方式的选择 本试验以甲醇:0.03 mol/L磷酸(含0.2%三乙胺)=80:20作为流动相系统进行等度洗脱,只出现一个峰,且前沿峰明显,产生的原因可能是柱温低、样品溶剂使用不当、柱过载等。采用梯度洗脱甲醇20%~50%2峰仍未分开,最后采用20%~60%甲醇梯度洗脱终使2峰完全分开。
2.6.3 流动相的组成及选择 2005年版药典中以水:乙腈:三乙胺=799:200:1(用氢氧化钠试液或冰醋酸调节pH值至5.9)为流动相,等度洗脱。为提高方法环境适用性,本试验考察了甲醇:水:冰醋酸:三乙胺 =225:345:0.5:0.2,作为流动相体系,虽然SMZ和TMP可以得到分离,但保留时间不一致;而采用甲醇:0.03 mol/L磷酸(含0.2%三乙胺)=80:20作为流动相等度洗脱,只出现溶剂峰,样品未分离;最后采用甲醇20%~60%梯度洗脱,能使2峰达到基线分离、保留时间一致、分离度好、重现性好、结果准确,可作为该片剂质量控制的参考方法。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:化学工业出版社,2000.
[2]聂仲伦,张传海,董玉亮.倍率减差双波长分光光度法测定复方磺胺甲恶唑片的含量[J].中国医院药学杂志,1996,16(7):292~294.
[3]刘承叶.三波长分光光度法测定复方新诺明片含量[J].药物分析杂志,1986,6(4):213~214.
[4]阮俊.吸收度线性组合法测定复方磺胺甲恶唑片含量[J].药物分析杂志,1991,11(5):289~291.
[5]孙增先,淡恒山.双波长比值光谱法测定复方磺胺甲恶唑片含量[J].中国医院药学杂志,1999,19(9):536~539.
[6]胡军.系数倍率双波长紫外光谱法测定复方磺胺甲恶唑片含量[J].医学文选,2001,20(4):473.
[7]朱碧军,程奇珍,叶久之.HPLC法测定复方磺胺甲恶唑片中磺胺甲恶唑与甲氧苄啶的含量[J].中国药品标准,2002,3(6):43~44.
[8]安登魁.药物分析[M].济南:济南出版社,1992.
[9]卫生部药政局.中国医院制剂规范[M].北京:中国医药科技出版社,1995.
G424.31
B
1671-1246(2011)08-0110-02
