碳化硅表面氢气吸附机理研究
2024-02-01尹博闻曾庆丰刘建涛刘永胜张相华
尹博闻, 曾庆丰, 关 康, 王 璐, 刘建涛, 刘永胜,董 宁, 张相华, 彭 诚
(1.西北工业大学 材料学院 超高温结构复合材料重点实验室, 西安 7100721; 2.华南理工大学 材料科学与工程学院,广州 511442; 3.西南交通大学 机械工程学院, 成都 610031; 4.西北核技术研究所,西安 710024)
1 引 言
SiC(碳化硅)材料具有宽禁带、高击穿场强、高热导率以及高载流子饱和漂移速度等,使其更适合于高频率、大功率、高温的半导体器件和短波长光电器件,成为当前国际半导体器件研究与产业化的热点[1]. Si-C共价键的高温稳定性以及碳化硅的自钝化特性使其还能作为重要的高温热结构材料[2]. 此外,SiC以其低热中子吸收截面和优良抗辐射性被广泛应用于核反应堆燃料的包覆材料[3]. 以上应用中,SiC多以薄膜或涂层的形式存在. 其制备过程通常以H2为载气,将含有Si、C元素的气体(如SiCl4、CH4、Cl3CH3Si等)输入反应器中,混合气体在基片上发生化学气相沉积(Chemical Vapor Deposition,CVD)制备而得. 在CVD过程中,H2和沉积的SiC会发生表面反应,这些表面反应会影响沉积产物SiC的表面品质及其应用性能,因此研究这些反应的具体过程及内在机理具有十分重要的科学意义和工程价值[4]. 上述研究大多基于实验探索,如通过改变宏观工艺条件来观测SiC的沉积速率和形貌,但这很难揭示其微观反应过程及量化沉积机理. 通过文献调研发现,吴广新等人[5]采用第一性原理计算模拟方法研究了H原子在Mg(0001)表面的吸附、扩散等现象,获得了大量理论数据. 在Mg(0001)表面上,H原子最稳定的吸附位为fcc(面心立方位),计算结果和实验吻合. 陈建辉等人[6]研究了氧在Nb(110)表面的吸附行为,表明O在最稳定的“洞位”随着覆盖率的增大吸附能呈现下降的趋势,说明O和O之间的排斥作用增加,吸附能减小. 本研究从微观原子角度出发,考虑能量、电荷、化学键等方面,研究气体分子从解离、扩散、吸附到发生进一步表面反应(脱附、腐蚀)的过程,分析其与SiC表面反应的微观机理.
2 计算模型和方法
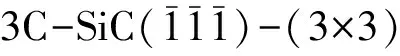

图1 (a)OT;(b)BR;(c)H3;(d)T4四种位置优化前结构,其中白色为H原子,黄色为Si原子,灰色为C原子Fig. 1 (a)OT;(b)BR;(c)H3;(d)T4 four positions optimized prestructure,of which white is H atom,yellow is Si atom,and gray is C atom
本文所涉及的计算均采用密度泛函软件包CASTEP,k点设置为3×3×1可得到收敛的能量值,交换关联能采用广义梯度近似的GGA-PBE[8];实空间截距为4.0 Å;精确的布里渊区采样在3×3×1的Monkhorst-Pack格子中进行,对应的抽样间距为0.05 Å-1;平面波展开的截断能设置为400 eV,对所有元素采用全电子基组;能量、梯度和位移的收敛标准分别为:2×10-5Hartree、4×10-3Hartree/Å和5×10-3Hartree/Å[9].
3 计算结果与讨论
3.1 H和H2的稳定吸附位
首先将氢原子分别放置在SiC表面OT位、BR位、H3位和T4位并进行结构优化,得到四种位置优化后的结果. 如图2所示,四种位置经结构优化后都为OT位,说明OT位为H吸附的最稳定位点,该位置吸附能最高,其余三种吸附位无法稳定存在.
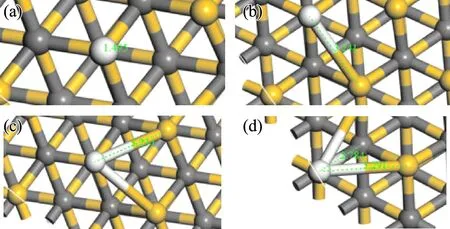
图2 (a)OT;(b)BR;(c)H3;(d)T4四种位置优化后结构,其中白色为氢原子,黄色为Si原子,灰色为C原子Fig. 2 (a)OT;(b)BR;(c)H3;(d)T4 has four positionally optimized structures,where white is hydrogen atom,yellow is Si atom,and gray is C atom
接着将H2分别放置于上述四种位点进行吸附,四种结果如图3所示,可以看出四种初始吸附位下H2均会进行自发解离,H-H键断裂,两个H原子均以OT位形式吸附在Si终端. 这一结果表明:H2在适当高度时会发生与初始吸附位点无关的自发解离性吸附,根据吸附能结果可进一步验证其属于稳定的化学吸附;双OT位的结果与H原子单独吸附时的稳定吸附位一致,这表明H2解离后两H原子之间的相互作用不足以影响H原子各自的吸附结果.
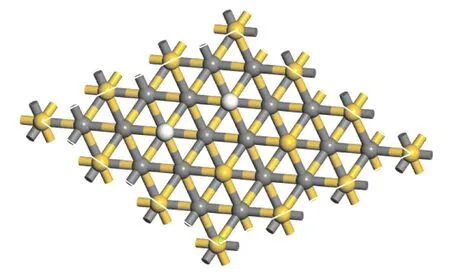
图3 H2分子优化后吸附结果,其中白色为H原子,黄色为Si原子,灰色为C原子Fig. 3 The adsorption results after optimization of H2 molecules,where white is H atom,yellow is Si atom,and gray is C atom
3.2 H2分子吸附后体系的电荷布居、键布居分析
3.2.1电子局域函数(ELF)分析
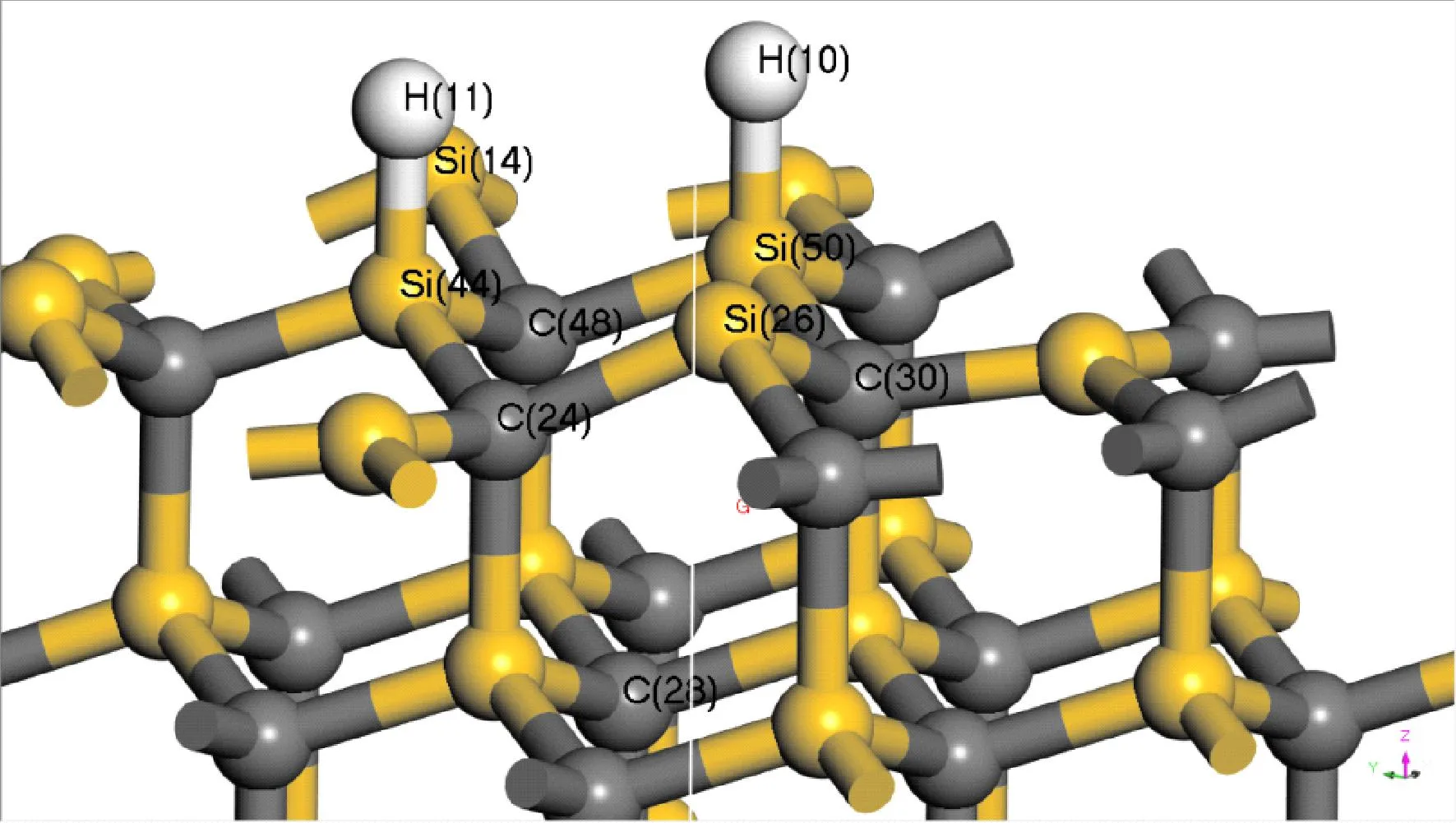
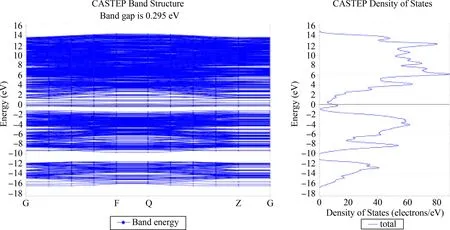
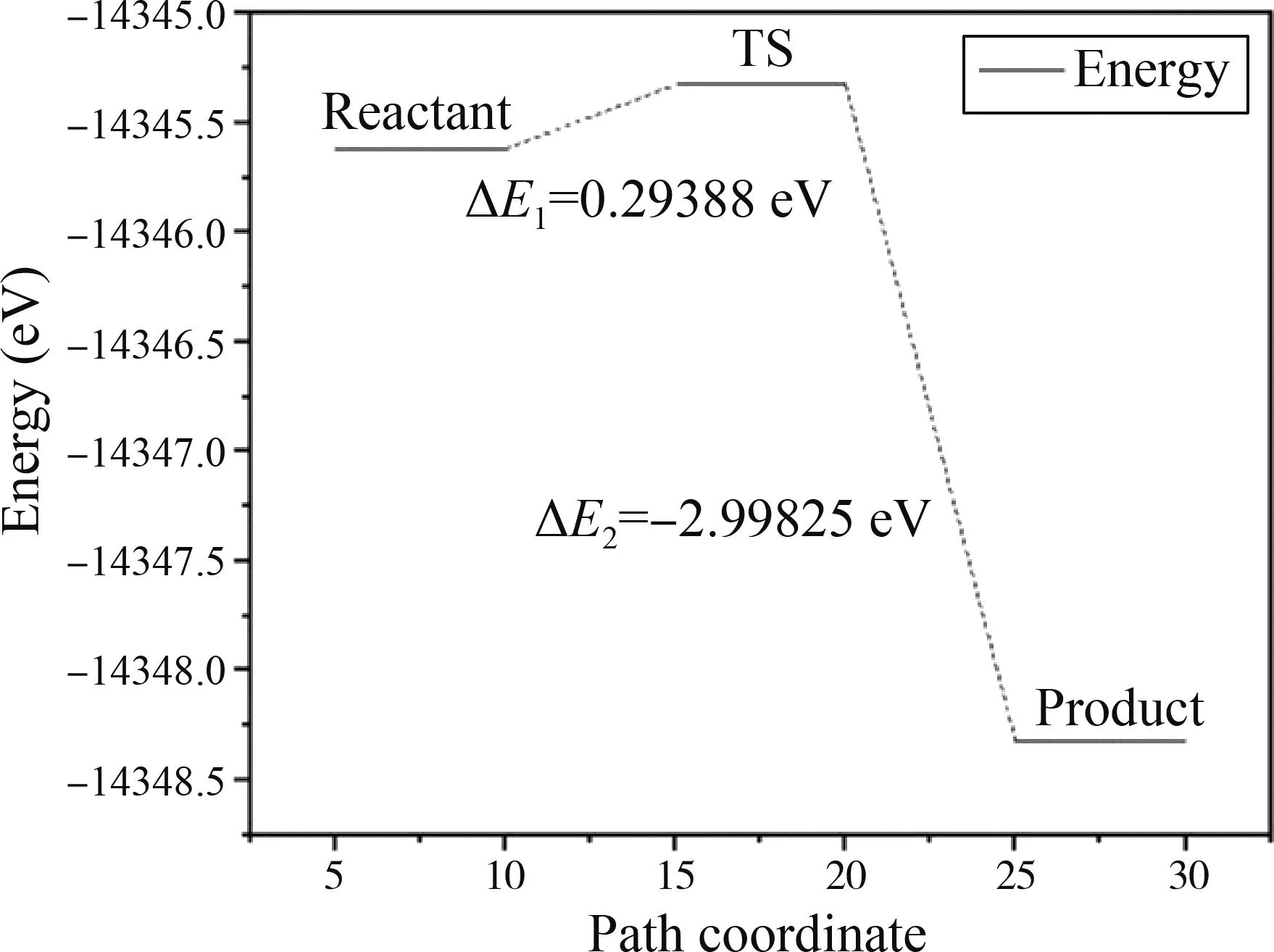
电子局域函数(Electron Localization Function,ELF)分析可以定性地判断成键特性. ELF值域为0~1,当0.75 图4 氢气吸附3C-SiC(111)表面差分电荷密度图Fig. 4 Differential charge density diagram of hydrogen adsorption 3C-SiC(111) 3.2.2电荷布居分析 表1给出了两个H原子及周围的Si和C原子在吸附前后的电荷转移情况. 可以看出:两H原子各得到0.09 eV电子,与之相连的Si原子失去0.26 eV电子,而下层的C原子几乎不发生变化. 即从Si原子向H原子发生了电荷转移,与上述差分电荷密度图所示基本一致. (H、Si、C原子的序号示意图如图5所示) 表1 H、Si、C原子在吸附前后电荷转移量 图5 氢、硅、碳原子的序号示意图Fig. 5 Schematic diagram of the serial number of hydrogen,silicon and carbon atoms 如图6所示为吸附后3C-SiC(111)表面的能带结构与DOS图,带隙为0.295 eV. 从图中可以看出,价带被分割成两部分,即上价带与下价带[11]. DOS图中显示费米能级处有一定的电荷密度,导带处存在多个能态密度峰,价带区域存在上下两个峰区,与能带结构图相对应. 图6 3C-SiC(111)表面的能带结构与DOS图Fig. 6 3C-SiC (111)surface of the band structure and DOS diagram 图7(a)与(b)分别为与H原子相连的Si原子、下层C原子以及H原子吸附前后的PDOS图像,由图可知,H原子在吸附后产生了多个峰值,且费米能级处产生电荷密度;Si原子在吸附后费米能级处的峰值向左移动,主要为s轨道和p轨道共同贡献;C原子由于在下层,在H原子吸附前后整体的峰值及轨道几乎未发生变化,再次证实了从Si原子向H原子发生了电荷转移. 通过计算吸附能与结构整体所吸附气体原子的个数关系,可以探究随着吸附原子个数的增加,整体稳定性的变化. 查找文献所知,吸附能的计算方法如式(1)所示[12]: (1) 其中Ead为吸附能,E是SiC表面吸附H原子后的总能量,E0为SiC表面未吸附H原子时的总能量,N为H原子的个数,E(X)为单个H原子的能量. 图8 3C-SiC(111)表面与表面覆盖率与吸附能之间的关系图Fig. 8 Relationship between 3C-SiC(111)surface coverage and surface coverage and adsorption energy 若吸附能为负值,则为放热反应,绝对值越大说明整体的稳定性越好,末态能量越低,处于一个相对稳定的状态;若为正值,则为吸热反应,绝对值越大说明整体的稳定性越差,末态能量越高,处于一个相对不稳定状态[14]. 由此可知,整体吸附能为负值,为放热反应,当覆盖率(θH≤4/9 ML)时,随着H原子的增加,H原子之间存在着较强的互相吸引作用;当覆盖率(θH>4/9 ML)时,此时H原子数目过多,吸附的H原子之间开始产生了部分不利于结构稳定的排斥力,使得吸附能的增长幅度减缓. 综上所述,随着覆盖率的增大,吸附能不断增大且整体为放热反应,H原子数目的增多会使结构趋于更稳定的状态. 过渡态是指反应物体系转变成产物体系过程中,经过的能量最高状态(或称活化络合物). 过渡态键的状况是:旧键未完全断裂,新键未完全形成. 过渡态是不稳定的,不能分离出来. 过渡态和反应物的能量差(△E)称为活化能[15]. 不同的反应体系有不同的活化能,活化能越大反应越困难. 对氢分子吸附在3C-SiC(111)表面的“解离-扩散-吸附”过程进行了过渡态搜索. 由图9可知,初态与过渡态之间的能量差值ΔE1为0.29388 eV,为吸热反应;过渡态与末态之间的能量差之ΔE2为-2.99825 eV,为放热反应;初末状态总能量差值ΔE为-2.70437 eV,此过程能量降低,总过程为放热反应. 图9 3C-SiC(111)表面H2分子吸附过渡态搜索的反应能量路径图Fig. 9 Reaction energy path diagram of the search for the transition state of H2 molecule adsorption on the surface of 3C-SiC(111) 图10为过渡态搜索三个过程中3C-SiC(111)表面和H2分子的状态. 由图9和图10可知,H2分子在3C-SiC(111)表面吸收热量开始解离达到过渡态,再释放热量达到末态,最终为两个分别吸附在3C-SiC(111)表面OT位的H原子,与3.1节描述相符. 图10 3C-SiC(111)表面H2分子“解离-扩撒-吸附过渡态”结构图:(a)初态结构;(b)过渡态结构;(c)末态结构Fig. 10 Structure diagram of the "dissociation-amplification-adsorption transition state" of H2 molecules on the surface of 3C-SiC(111):(a)initial structure;(b)transitional structures;(c)terminal structure (1)H原子吸附在SiC表面时,主要以OT(顶位)的方式存在且吸附能较高,可稳定存在,其余初始吸附位最终均会移动到OT位. (2)H2分子在四种吸附位均会产生自发的解离性吸附,最终两H原子以双OT位形式稳定吸附在Si终端. (3)从电荷布居图和差分电荷密度图中可以看出,吸附的两H原子得到电子,而与之相连的Si原子失去电子,电荷从Si原子向H原子转移,下层的C原子由于距离较远,几乎未发生电荷转移,其PDOS图几乎不发生变化. (4)随着H原子在表面覆盖率的增大,吸附能逐渐增大,结构处于更加稳定的状态,在覆盖率小于4/9时H原子有很强的相互吸引力,随着H原子数目增多,排斥力逐渐增大,吸附能增长幅度减少. (5)过渡态搜索可以发现,在解离的过程中会先升高能量成为一个过渡状态,而后能量下降变为解离的最终状态(OT位),且能量变化不大. 致 谢:感谢西北工业大学高性能计算中心提供的计算服务.

3.3 能带结构分析
3.4 吸附能与覆盖率之间的关系

3.5 氢分子解离与扩散的过渡态搜索
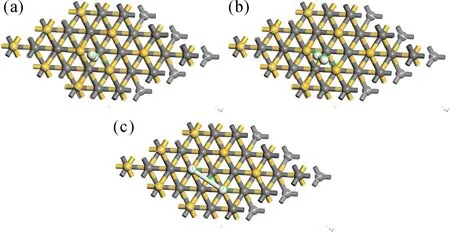
4 结 论
