dsDNA与TiO2(110)在溶液中相互作用的动力学行为和机制
2024-02-01胡书环孟范超
杨 晋, 胡书环, 刘 雯, 孟范超
(1. 中国科学院大学 生命科学学院, 北京 100049; 2. 深圳华大生命科学研究院, 深圳 518083; 3. 广东省高通量测序工程技术研究中心, 深圳 518083; 4. 康年医疗科技有限公司,山东 烟台 264030; 5. 烟台大学 精准材料高等研究院, 烟台 264005)
1 引 言
二维过渡族金属氧化物(transition metal oxide,TMO)主要包括TMO的单层结构、TMO的多层结构和不同种类TMO的多层和超晶格结构[1]. 对于二维纳米材料,由于表面性能控制着材料的行为,二维TMO通常表现出与其块体显著不同的物理和化学性质,如高温超导性和多铁性,以及独特的光学、力学和热学性质[1]. 目前,许多二维TMO,例如MoO3、V2O5、TiO2、ZnO等[2-4],已经被报道可以通过分层或自底向上生长的方式可控制备,而且具有优异的化学和热稳定性[5,6]. 近年来,二维TMO在光学、电子、催化、能源装置(电池、超级电容器、太阳能电池和燃料电池)、传感器和生物系统等方面已经实现应用或者具有潜在的应用前景[1]. 由于本文的研究重点是生物传感,因此,特别值得指出的是,二维TMO通常具有较高的等电点,可以通过静电相互作用固定一系列具有低等电点的生物分子,例如皮质醇和多巴胺,并且能够促进吸附体系之间的电荷转移[7]. CuO、WO3、TiO2和ZnO等都展现出了相当低的检测限值[8-11].
本文作者[12]前期通过第一性原理计算发现,二维TMO中典型的金红石型TiO2纳米层(110)表面对DNA的四个碱基具有强吸附性能,其吸附能比石墨烯及其衍生物的吸附强度大两倍以上. 在实际应用中,DNA通常是在溶液中与表面相互作用. 然而,第一性原理计算受限于有限的原子数量,无法用于研究长链DNA与TiO2(110)表面在溶液中的相互作用行为和机制. 在此方面,基于原子力场的分子动力学模拟常被用于研究吸附质和吸附剂在溶液中的吸附行为. 例如,Brandt和Lyubartsev[13]利用分子动力学模拟研究TiO2(100)表面与氨基酸类似物的相互作用发现,TiO2(100)与具有极性和芳香侧链的氨基酸之间的相互作用强于具有甲基的氨基酸. Mao等[14]使用分子动力学模拟研究了TiO2(100)表面的丝氨酸磷酸化效应,发现与TiO2(100)表面结合的肽的结合自由能随着磷酸化而增加. Liu等[15]使用分子动力学模拟研究了DNA在二维MoS2表面的吸附取向,发现DNA倾向于沿轴向垂直吸附到MoS2表面. 金红石型TiO2(110)是TiO2自然存在的最稳定表面,然而,迄今为止,其与DNA在溶液中相互作用的行为与机制仍有待进一步研究.
因此,本研究采用分子动力学模拟,研究dsDNA与金红石型TiO2(110)表面在溶液中的相互作用行为和机制. 本文首先构建了dsDNA在溶液中轴向初始平行或垂直于TiO2(110)表面这两种计算模型. 进而,通过能量最小化和长时结构弛豫,研究了dsDNA在吸附过程中的稳定吸附构型、动力学行为和结构稳定性. 最后,通过计算二者之间的相互作用能和水分子密度/数量,讨论了dsDNA在TiO2(110)表面的吸附机制.
2 计算方法
本文的分子动力学模拟利用GROMACS (Groningen Machine for Chemical Simulation)软件包的2022.2版本完成[16]. 初始模拟体系的超晶胞如图1所示,包含水分子、Na+离子、dsDNA和具有(110)表面的金红石型TiO2纳米层. dsDNA包含16对碱基,共1014个原子,碱基排序为5’-ATCGATCGATCGATCG-3’(标号为1-16)和3’-TAGCTAGCTAGCTAGC-5’(标号为32-17). A、T、C、G分别代表腺嘌呤、胸腺嘧啶、胞嘧啶、鸟嘌呤这四种由氮碱基、脱氧核糖和磷酸组成的脱氧核苷酸分子. TiO2(110)表面模型共包含11880个原子. DNA在初始时沿轴向平行或垂直置于TiO2(110)表面上方,并且保持二者之间的最小间距为1.5 nm. 为了便于标记,如图1a和1b所示,将两种初始构型分别标记为DNA ∥ TiO2(110)和DNA ⊥TiO2(110). 图1a和1b的模型中分别包含30个Na+离子以平衡DNA的电负性以及27671个和27667个水分子. 因此,图1a和1b模型的总原子数分别为95937个和95925个. 超晶胞具有完全周期性,且在X×Y×Z的尺寸分别为9.80 nm × 9.87 nm × 10.32 nm,其中Z定义为垂直于TiO2(110)表面的方向.
在分子动力学模拟中,DNA采用CHARMM 27力场[17]. TiO2采用Brandt和Lyurbatsev开发的用于模拟TiO2与水相互作用的原子力场参数[18],如表1所示. 该力场已被用于TiO2(100)表面与氨基酸类似物的相互作用[13],TiO2(100)表面的丝氨酸磷酸化效应[14],和TiO2纳米球与脂质双层黏附[19]等研究中. 水分子采用TIP3P模型. 在模拟过程中,首先约束DNA的位置,采用最速下降算法对超晶胞进行能量最小化,时间步长为1 fs,收敛标准为作用力小于1000 kJ/mol/nm. 随后,保持DNA的位置约束,使用等温系综对超晶胞进行100 ps平衡,时间步长为1 fs. 最后,解除对DNA的位置约束,对超晶胞进行200 ns的平衡模拟,时间步长为2 fs. 在上述模拟中,选取以下共性模拟参数. 短程静电和范德华相互作用的截断距离为1.2 nm. 恒温器采用V-rescale使模拟系统的温度维持在300 K,时间耦合常数设置为0.1 ps. 采用LINCS算法约束所有H键. 长程静电相互作用使用PME. 此外,在整个模拟过程中,保持TiO2中所有原子的位置不变. 模拟结构的视图通过VMD软件获得[20].
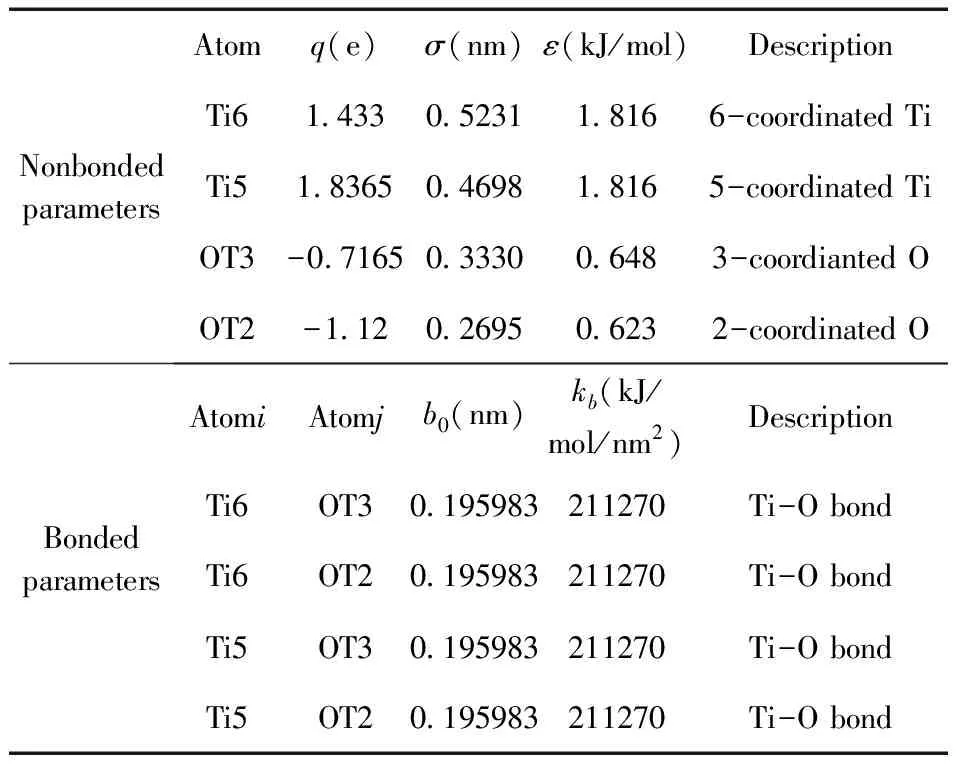
表1 TiO2 (110)力场的非键和键合力场参数
3 结果和讨论
3.1 dsDNA在TiO2 (110)表面的吸附行为
DNA ∥ TiO2(110)和DNA⊥TiO2(110)这两种体系经过200 ns弛豫后的结果如图2所示. 可以看到,对于这两种初始构型,经过弛豫后DNA均平行于TiO2(110)表面,呈现出相同的吸附特征. 此外,大部分Na+离子分布于DNA的周围. 根据以往研究报道,DNA在氮化石墨烯[21]、二硫化钼[15]、二硒化钼[22]等二维表面的稳定吸附构象为DNA主轴与吸附表面近乎垂直. 以二硒化钼为例,位于表面的硒原子带有负电荷,而DNA也呈负电性,因此二者之间的电荷排斥导致DNA无法通过外侧的负电荷磷酸基团平行吸附于二硒化钼表面. 然而,垂直吸附势必通过破坏DNA双链碱基对之间的氢键结构以使末端碱基与表面形成吸附,不利于DNA的结构稳定性. 不同于以上研究,本研究发现DNA的稳定吸附构象为平行吸附于TiO2(110)表面. 这可能归因于TiO2(110)表面既有带负电荷的OT3和OT2原子,又有带正电荷的Ti5原子(如表1所示). 水平吸附有助于减少对DNA结构的破坏. DNA结构的稳定性将在下文3.2节部分进行详细论述. 此外,DNA吸附的机制将在下文3.3节中展开.
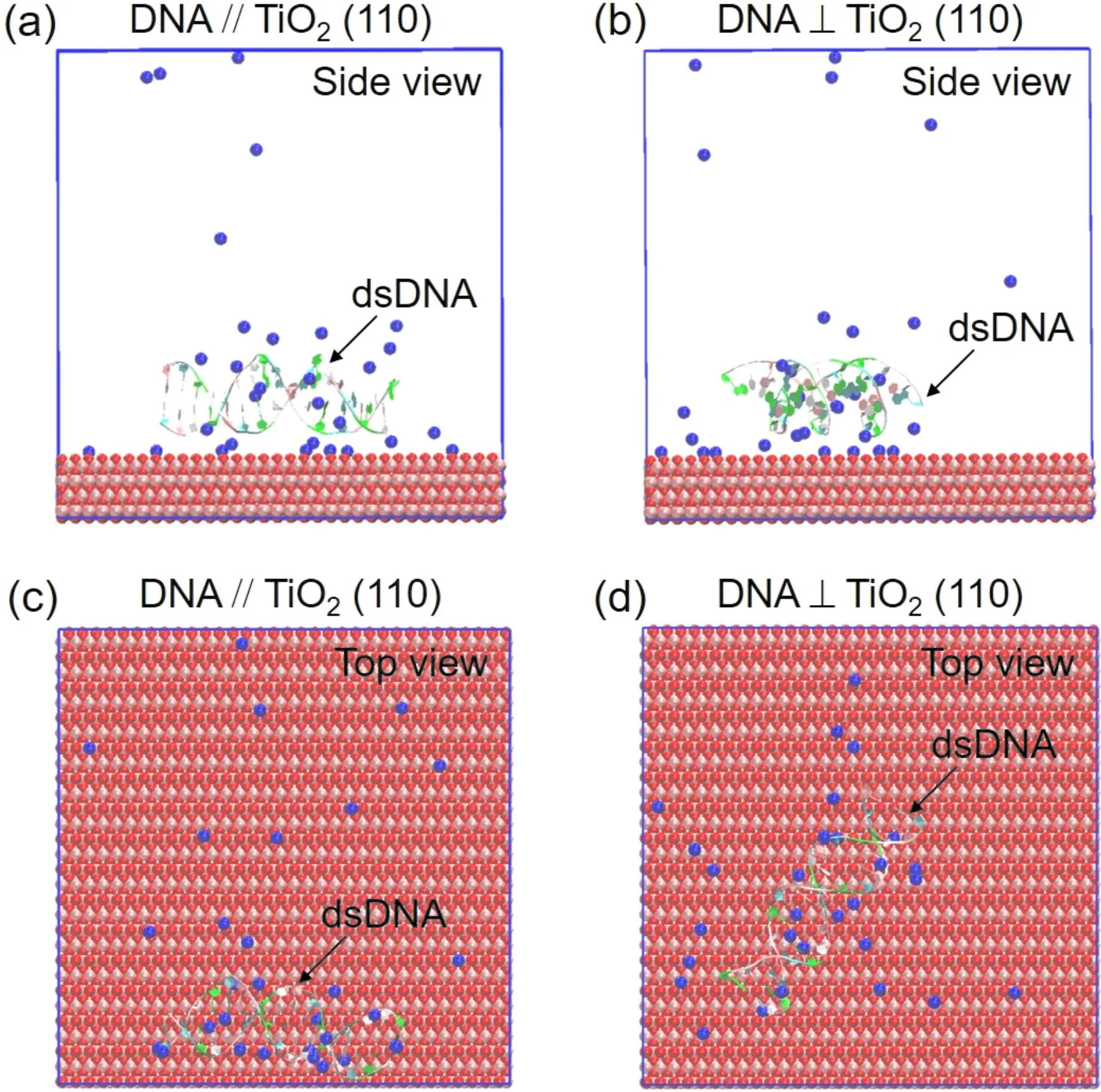
图2 经过200 ns弛豫后,DNA∥TiO2 (110)和DNA⊥TiO2 (110)体系的侧视(a-b)和俯视(c-d)图Fig. 2 The side (a-b)and vertical (c-d)views for DNA∥TiO2 (110)and DNA⊥TiO2 (110)systems after a 200 ns relaxation
为了进一步研究DNA在TiO2(110)表面吸附的动力学过程,图3a和3b分别绘制了吸附到TiO2(110)表面的DNA原子数随弛豫时间的演变. 注意,与TiO2(110)表面间距小于0.6 nm的DNA原子被定义为吸附原子. 对于DNA ∥ TiO2(110)初始体系,吸附原子数量呈现两个阶段性的增加. 在42 ns时,DNA首次被吸附到TiO2(110)表面,吸附原子数约为10,并保持到65 ns. 随后,在66 ns,吸附原子数开始剧烈增加,并稳定到40 ~ 50个原子数的区间. 而对于DNA⊥TiO2(110)初始体系,吸附原子数展现为三个阶段,包括24 ns时首次跃升到10个原子左右,80 ns增加到20个原子左右,以及118 ns以后在40 ~ 50原子区间内波动. 从整体上看,初始平行或垂直于TiO2(110)表面放置DNA不影响最终吸附的DNA原子数量.
进而,以DNA ∥ TiO2(110)初始体系为例,研究了DNA在TiO2(110)表面吸附过程中的构型变化,如图4所示. 通过观察发现,在50 ns时,T32中的原子首先吸附到了TiO2(110)表面. 随后,对比DNA在50 ns和60 ns的构型,可以看出整个DNA似乎以T32为中心呈现出向TiO2(110)表面翻转吸附的趋势. 进而,在70 ns,DNA完成在TiO2(110)的吸附,吸附的DNA碱基包括一条单链中的A5、T6、C15、G16和另一条单链中的G22、A23、A31、T32,即TiO2(110)表面同时吸附了四种碱基. 从70 ns至200 ns,DNA的吸附构型没有明显变化. 因此,可以推断,DNA在TiO2(110)表面的吸附强度和稳定性较好. 此外,这些关键时刻的构型变化与图3a中DNA吸附原子数量的变化相一致.
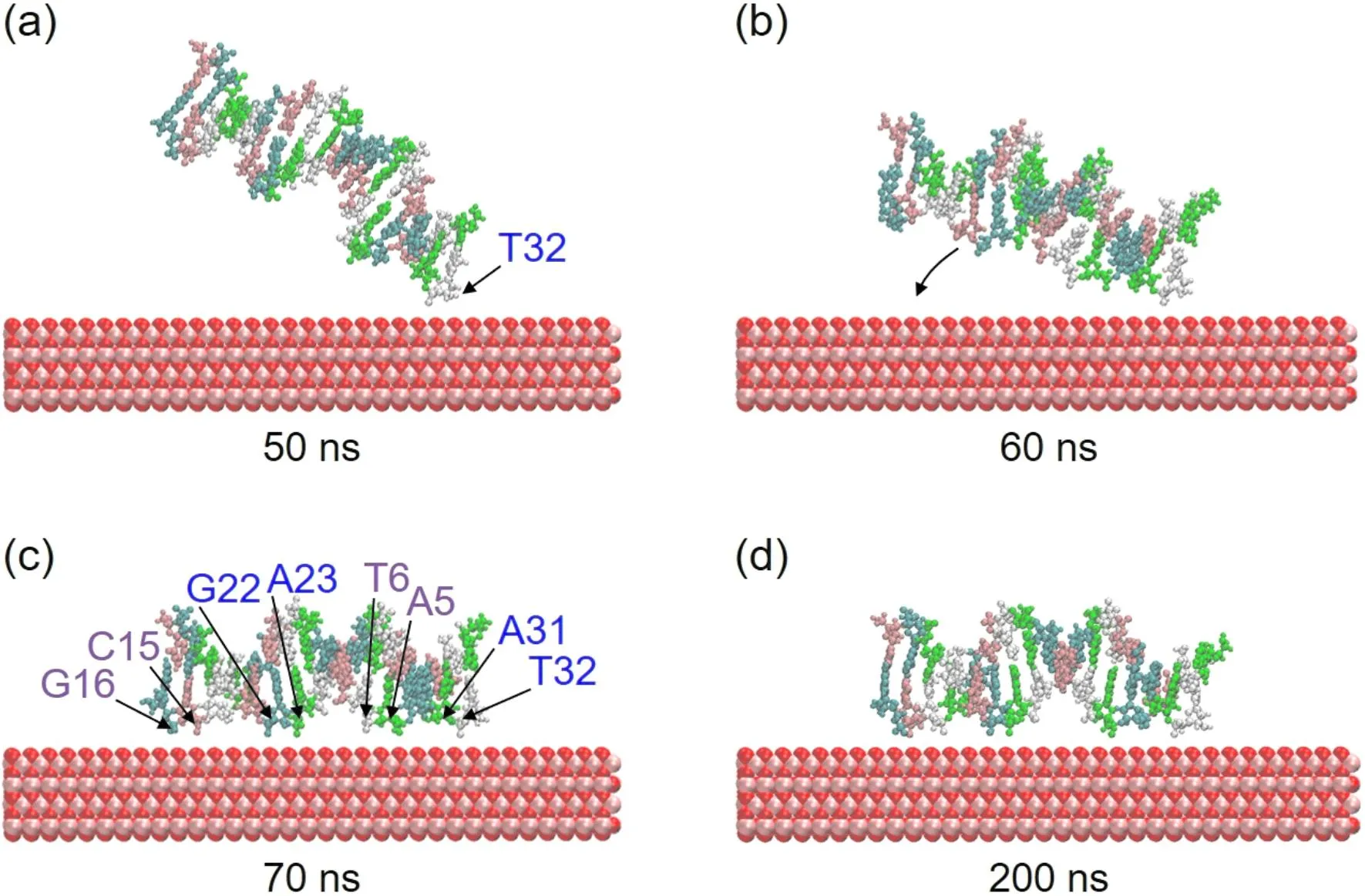
图4 DNA ∥ TiO2 (110)初始体系在(a)50 ns、(b)60 ns、(c)70 ns和(d)200 ns模拟时刻下的DNA和TiO2 (110)吸附构型Fig. 4 The configurations of DNA and TiO2 (110)at simulation time of (a)50 ns,(b)60 ns,(c)70 ns,and (d)200 ns for DNA ∥ TiO2 (110)system
3.2 dsDNA在吸附过程中的结构稳定性
由于水平吸附不需要破坏DNA碱基之间的氢键完成吸附,因此可以推断TiO2(110)表面吸附DNA对其结构稳定性影响较小. 为探究TiO2(110)表面吸附DNA时对其结构的影响,计算了DNA碱基对间的氢键数量和DNA原子位置的均方根偏差(root mean square deviation,RMSD). 如图5所示,对于DNA ∥ TiO2(110)和DNA⊥TiO2(110)这两种体系,氢键数量随模拟时间的演变曲线基本重合,其均值和标准偏差分别为40.74 ± 1.35个和40.61 ± 1.45个. 并且在200 ns的模拟时间内,氢键数量基本没有变化. 这说明DNA在吸附到TiO2(110)表面时,双螺旋结构几乎没有受到影响. 此外,这两种体系的RMSD随着模拟时间的演变也基本一致. 并且RMSD的数值很小. 在200 ns时间内二者RMSD的均值和标准偏差分别为0.18 ± 0.03 nm和0.20 ± 0.04 nm. 因此,水平吸附有助于保持DNA结构的稳定性,并且金红石型TiO2(110)可以用作DNA的良好吸附剂.
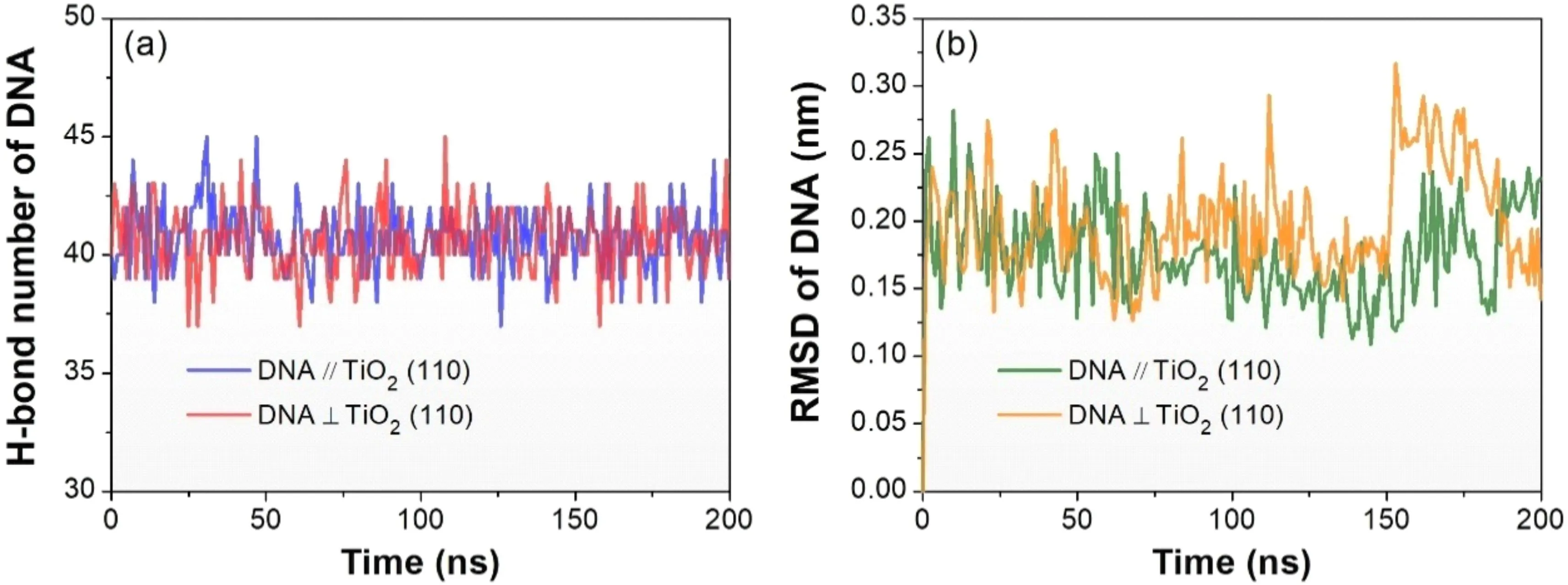
图5 DNA ∥ TiO2 (110)和DNA⊥TiO2 (110)体系的(a)DNA中的氢键数量和(b)DNA中原子位置的均方根偏差随弛豫时间的演变Fig. 5 The evolutions of (a)the number of H-bonds and (b)RMSD of DNA with respect to simulation time for DNA ∥ TiO2 (110)and DNA⊥TiO2 (110)systems
3.3 dsDNA在TiO2 (110)表面的吸附机制
为了探究DNA分子在TiO2(110)表面的吸附机制,研究了两种初始吸附体系的DNA与TiO2(110)之间的短程范德华相互作用能,如图6a和6b所示. 通过对比图3a和6a以及图3b和6b,可以明显看出,二者之间相互作用能的阶梯式演变与DNA吸附原子个数的阶梯变化具有强相关性. 这表明范德华相互作用在DNA吸附过程中起着重要作用. 此外,对于水平和垂直初始吸附体系,范德华相互作用能均稳定在-150 kJ/mol左右,这也与两种体系在弛豫末期吸附的DNA原子个数相当的结论相一致. 值得指出的是,在使用PME的静电作用下,库伦静电相互作用能无法针对相互作用对进行分离. 本文尝试通过使用Cut-off方法代替PME并对模拟进行重计算,发现DNA和TiO2(110)间的远程静电相互作用能可达约-12000 kJ/mol. 尽管将相互作用能针对DNA和TiO2(110)进行分解的结果不一定真实,DNA在TiO2(110)表面的水平吸附可能暗示远程静电相互作用对吸附起着至关重要的作用.
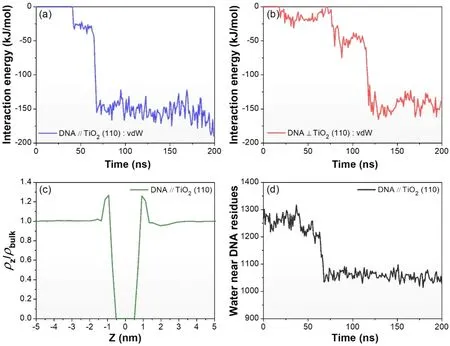
图6 (a)DNA ∥ TiO2 (110)和(b)DNA⊥TiO2 (110)体系中DNA和TiO2 (110)短程范德华相互作用能随弛豫时间的演变. (c)沿Z方向的水密度分布. (d)分布在已吸附碱基,包括A5、T6、C15、G16、G22、A23、A31、T32,周围的水分子数量随弛豫时间的演变Fig. 6 The short-range van de Waals (vdW)interaction energies as a function of simulation time for (a)DNA ∥ TiO2 (110)and (b)DNA⊥TiO2 (110),respectively. (c)is the relative water density to the bulk water along the Z direction. (d)shows the number of waters near the contacted DNA residues,including A5,T6,C15,G16,G22,A23,A31,T32,with respect to the simulation time
由于溶液中水分子对生物分子的吸附也起着重要作用[23,24],因此,以DNA ∥ TiO2(110)体系为代表,图6c和6d研究了水分子沿Z轴的密度分布以及在已吸附DNA碱基1.0 nm范围内水分子数量随模拟时间的演变. 如前所述,对于DNA ∥ TiO2(110)体系,这些吸附的碱基包括A5、T6、C15、G16、G22、A23、A31、T32. 从图6c中可以看出,TiO2(110)表面与水分子存在色散相互作用,使得水分子在距离TiO2(110)表面约1.0 nm的位置处出现一个密度峰值. 因此,第一溶剂化水层位于TiO2(110)表面间距1.0 nm处,其密度约为体相水密度的1.26倍. 进而,随着DNA碱基逐渐吸附到TiO2(110)表面,如图6d所示,在50 ns左右时,与TiO2(110)表面接触的碱基周围的水分子出现明显下降,约有200个水分子从TiO2(110)表面解除吸附. 水分子下降的时刻也与图3a和图4a中碱基开始吸附的时刻基本一致. 这说明在DNA的吸附过程中,第一溶剂化水分子层中的水分子被DNA挤压,产生纳米尺度的“脱湿”过程. 纳米级脱湿为DNA的吸附提供了驱动力,进一步增强了DNA的吸附.
4 结 论
1)本文利用分子动力学模拟研究了dsDNA与金红石型TiO2纳米层(110)表面在溶液中相互作用的动力学行为和机制.
2)通过对dsDNA吸附后构型、吸附动力学过程和结构稳定性的分析发现,在初始时沿轴向平行或垂直于TiO2(110)表面放置的dsDNA的稳定吸附构型均为水平吸附. 水平吸附不仅使得dsDNA的四种碱基均吸附到TiO2(110)表面,增加了吸附稳定性,而且不破坏dsDNA的结构稳定性.
3)对dsDNA和TiO2(110)之间相互作用能和水分子密度/数量的分析发现,水平吸附可能源于二者之间的短程范德华和长程静电相互作用. 此外,纳米尺度脱湿进一步增强了dsDNA的吸附.
4)本研究探明了dsDNA与TiO2(110)表面在溶液中相互作用的动力学行为和机制,为DNA传感和测序等应用指出了一种非常有潜力的材料.
5 数据可用性
支持本研究结果的数据已存入中国国家基因库数据库(CNGBdb)[25]的CNGB序列档案库(CNSA)[26],登录号为CNP0003376.
致 谢:感谢国家基因库(China National GeneBank,CNGB)和广东省高通量测序工程技术研究中心(Guangdong High-throughput Sequencing Research Center)对本项目的支持.
