Si,Ge,Zr和Sn掺杂SrTiO3的电子结构和光催化性能第一性原理研究
2024-01-18熊明姚孔维静杨淑敏
熊明姚,孔维静,胡 斌,杨淑敏
(喀什大学 物理与电气工程学院,喀什 844000)
1 引 言
由于持续的人类活动和工业化,水污染是一个全球性的严重问题,一直影响着人类社会的方方面面[1,2].从传统技术,如消毒和污水回收等,到先进技术,如半导体光催化和生物吸附等[3,4].近年来,钙钛矿型化合物被广泛用于光催化分解水.特别是,SrTiO3已被发现是该领域潜在的光催化剂之一[5,6].基于ABX3型晶体结构的铁电材料,由于其介电性能,在理论和实验研究中得到了广泛的研究,SrTiO3具有铁电性、半导体性和催化活性等物理特性,是非常重要的材料[7-10].
SrTiO3的电子和光学特性一直是研究人员的兴趣所在,在这方面,科研工作者们使用了不同的掺杂剂,提高了SrTiO3材料的性能.如龚凌云[11]等采用第一性原理研究了不同浓度的Nb掺杂SrTiO3体系的电子结构、光学性质和力学性质,发现SrTiO3、SrTi0.875Nb0.125O3和SrTi0.75Nb0.25O3三种体系的带隙分别为1.837 eV、1.924 eV和1.939 eV,掺杂使得SrTiO3体系的带隙呈现增大的趋势.薛卫东等[12]运用第一性原理计算了氧空位对SrTiO3薄膜电子结构的影响,氧空位导致SrTiO3体系的带隙提高了1.75 eV,由顺电转换为铁电.Yu等[13]采用溶胶凝胶水热法制备了可见光响应的Cr/N共掺杂SrTiO3体系,在Cr/N共掺杂SrTiO3的制备过程中,Cr的加入有利于N的掺杂,Cr/N共掺杂的SrTiO3光催化剂比Cr掺杂的SrTiO3具有更高的光催化制氢活性.Zhang等[14]采用基于密度泛函理论(DFT)计算研究了N掺杂、S掺杂和N/S共掺杂SrTiO3体系的电子结构和光学性质,N/S共掺SrTiO3的带隙值最小且可见光响应较高.Abdi 等[15]采用简单的固相反应路线,成功合成了La和Fe掺杂的SrTiO3纳米颗粒.研究了不同掺杂浓度(1、2、3、4和5 wt%)对制备样品特性的影响,所有样品都是立方钙钛矿结构,获得的4 wt%的La-Fe离子作为掺杂元素的最佳浓度,以实现96 %降解效率的高性能光催化活性.
所以,目前无论是从实验还是理论入手,SrTiO3在材料领域的都是研究热点.由于Si,Ge,Zr和Sn的一般价态与SrTiO3中的Ti相同且价格低廉,而且从目前的研究进展来看,还没有Si,Ge,Zr和Sn掺杂对SrTiO3性能影响的理论研究.因此,可用于SrTiO3材料的掺杂替代.在本文中,采用了QUANTUM ESPRESSO(QE)软件包来实现基于密度泛函理论(DFT)的第一原理计算,以探究Si,Ge,Zr和Sn掺杂SrTiO3的Ti位的结构、电子结构和光催化特性.
2 理论模型与计算方法
图1展示了文章采用的计算模型,由5个原子构成的SrTiO3体系为原胞,构建2×2×2的超胞,一共包括40个原子(8个Sr原子、8个Ti原子和 24个O原子),如图1(a)所示.分别采用一个Si,Ge,Zr和Sn原子取代超胞中的一个Ti原子,生成掺杂浓度为12.5 %的四种掺杂体系,如图1(b)所示,四种掺杂可以表示为SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn).
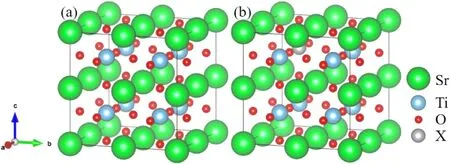
图1 SrTiO3的超胞模型(a)未掺杂的SrTiO3,(b)一个X原子掺杂的 2×2×2的SrTiO3超胞模型
文章所涉及的计算均通过基于密度泛函理论(DFT)的开源免费软件包QUANTUM ESPRESSO(QE)[16]实现.采用Perdew-Burke Ernzernhof (PBE)提出的广义梯度近似(GGA)用于处理交换相关能[17],截止能量为50 Ry(1 Ry=13.6056923 eV)[18].采用Monkhorst-Pack方案[19]进行布里渊区积分,其中选择3×3×3的k点进行自洽计算,非自洽计算使用6×6×6的k点.使用变胞弛豫方案,对五种体系进行优化,对于能带结构和态密度,采用QE软件包中包含的band.x和dos.x模块代码进行计算.
3 结果与讨论
3.1 几何优化结果
本征SrTiO3体系和SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系的晶格常数如表1中所示.本征SrTiO3的晶格常数计算值为3.928 Å,与实验值3.905 Å[20]非常接近,晶格常数的误差不到1 %.SrTi0.875Si0.125O3掺杂体系的晶格常数与本征SrTiO3的晶格常数相比略有下降,这与相关研究结果[21]一致,SrTi0.875Ge0.125O3晶格常数减小,其余两种掺杂SrTiO3体系的晶格常数增加.

表1 SrTiO3掺杂前后的晶格常数a,b,c,密度ρ,总能量E以及形成能Ef
为了检查四种掺杂结构的稳定性,根据公式(1)计算SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)的形成能Ef[22]:
(1)
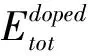
3.2 本征SrTiO3电子结构分析
在图2(a)中,显示了本征SrTiO3体系的能带结构,其中费米能级存在于0 eV.价带最大值出现在高对称点G点处,导带最小值也出现在G点处,这表明了G-G的直接带隙性质,其计算带隙为1.853 eV.这与SrTiO3的相关计算结果文献一致:带隙为1.838 eV的直接带隙[23],但低于3.2 eV[24]的实验结果,这主要是由于交换关联函数在使用GGA近似描述激发态时会低估体系的带隙值[25].然而,能带结构的带隙变化趋势仍然是可靠的.本征SrTiO3体系的总态密度和分波态密度如图2(b)所示,零点能量为费米能级.能量水平从价带的-5 eV到0 eV主要由O-2p轨道和Ti-3d轨道贡献,表现出明显的杂化现象,导带主要取决于Ti-3d轨道.能量范围在-20 eV至-15 eV主要由O-2s轨道和Sr-3p轨道杂化贡献.

图2 (a)本征SrTiO3的能带结构,(b)本征SrTiO3的态密度
3.3 Si,Ge,Zr和Sn掺杂SrTiO3的能带结构分析
计算了掺杂SrTiO3体系的能带结构,从能带结构的角度分析 Si,Ge,Zr和Sn掺杂对SrTiO3的影响.从图3可以看出,Si掺杂SrTiO3的带隙大小为1.849 eV且仍然为直接带隙,相比于本征SrTiO3体系的带隙有所减小,这与Liu等[21]的研究结果基本吻合.而对于SrTi0.875X0.125O3(X=Ge,Zr,Sn)三种掺杂体系而言,从图中可以看出Ge,Zr和Sn取代Ti位后,SrTi0.875X0.125O3(X=Ge,Zr,Sn)三种掺杂体系仍然具有直接带隙特征.而三种掺杂体系的带隙值相对于本征SrTiO3分别增加到1.916 eV,1.895 eV和1.925 eV.显然,Ge,Zr和Sn取代Ti位时对SrTiO3的带隙起到增大作用,Si取代Ti位起减小作用.

图3 能带结构:(a)SrTi0.875Si0.125O3;(b)SrTi0.875Ge0.125O3;(c)SrTi0.875Zr0.125O3;(d)SrTi0.875Sn0.125O3
3.3 Si,Ge,Zr和Sn掺杂SrTiO3的态密度分析
图4(a-d)显示了Si,Ge,Zr和Sn掺杂SrTiO3体系的分波和总的态密度图.Si掺杂后观察到SrTi0.875Si0.125O3的价带仍然主要由O-2p轨道和Ti-3d轨道贡献,导带主要由Ti-3d态贡献,Si对于总态密度的贡献不明显,SrTi0.875Si0.125O3的带隙相对于本征SrTiO3体系带隙减小.而对于SrTi0.875X0.125O3(X=Ge,Zr,Sn)三种掺杂体系可以明显的看出,对于导带部分,SrTi0.875Ge0.125O3体系的Ge-4s轨道,SrTi0.875Zr0.125O3体系的Zr-4d轨道和SrTi0.875Sn0.125O3体系的Sn-5s轨道,通过掺杂过程,都分别对自身的掺杂体系的总态密度加以贡献.对于本文研究SrTiO3中的Ti位被X(X=Si,Ge,Zr,Sn)原子取代.从总体上来看,对于SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系而言,除去SrTi0.875Si0.125O3体系,对于本征SrTiO3体系来说,三种掺杂体系的带隙变宽.也就是说,带隙遵循Sr8Ti7SiO24(1.849 eV) 图4 态密度:(a)SrTi0.875Si0.125O3;(b)SrTi0.875Ge0.125O3;(c)SrTi0.875Zr0.125O3;(d)SrTi0.875Sn0.125O3 为了描述本征SrTiO3和SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系相对于水氧化还原电位的带边,可以采用经验公式(2)和公式(3)得到半导体的价带带边的氧化电势(EVB)和导带带边的还原电势(ECB)[26].能带结构中GGA会低估带隙,尽管五种SrTiO3体系的结果不影响带隙的定性分析或电子结构等特性的研究,但是,在预测氧化还原电位和光吸收对应的半导体带边时,由于低估带隙而引入的误差将会变得很大.为了弥补这种低估,将引入“剪刀”算符这一方法.本文选用的剪刀算符的值为1.347 eV这是由本征SrTiO3的计算带隙值与实验值之间的差值决定的. ECB=X-EH-0.5Eg (2) EVB=Eg+ECB (3) 其中,X是本征SrTiO3和SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系对应的电负性.EH约为4.5 eV,是氢标度上自由电子的能量.Eg是五种SrTiO3体系对应剪刀算符的带隙值.EVB等于Eg和ECB之和. 由表2可以看出,计算出本征SrTiO3和SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系的EVB分别为:2.418 eV、2.460 eV、2.487 eV、2.442 eV和2.481 eV.而五种SrTiO3体系对应的ECB分别是-0.782 eV、-0.736 eV、-0.776 eV、-0.800 eV和-0.791 eV.图5给出了本征SrTiO3和SrTi0.875X0.125O3(X=Si,Ge,Zr,Sn)四种掺杂体系的带边位置.从氧化还原性质方面来看,本文包含的五种SrTiO3体系的EVB大于1.24 eV,ECB大于0.00 eV,均满足光解水的基本条件.其中,SrTi0.875Zr0.125O3体系的还原性最高,SrTi0.875Ge0.125O3体系的氧化性最高.SrTi0.875X0.125O3(X=Zr,Sn)两种掺杂体系的氧化还原性得到提高,而SrTi0.875Si0.125O3的还原性虽然降低,四种掺杂体系对于本征体系的氧化性都得到了提高. 表2 纯和掺杂SrTiO3的带隙能量(eV)(剪刀算符1.347 eV),电负性X,带边位置ECB和EVB. 采用QUANTUM ESPRESSO(QE)软件包,利用广义梯度近似(GGA)的第一原理模拟研究了Si,Ge,Zr和Sn掺杂SrTiO3的结构,电子结构和光催化性能.优化后的晶格常数值和实验值吻合较好.通过分波和总态密度解释了四种掺杂元素对能带结构的具体影响.掺杂后,除了SrTi0.875Si0.125O3体系的带隙值降低之外,SrTi0.875X0.125O3(X=Ge,Zr,Sn)三种掺杂体系的带隙值增加,费米能级保持不变,四种掺杂体系的导带底或者价带顶未出现穿过费米能级的现象,这可能是由于Si,Ge,Zr和Sn四种原子的一般价态与Ti的相等的缘故.基于相对于未掺杂SrTiO3的带边位置,获得了四种单掺SrTiO3体系的带边位置.对于掺Si的SrTiO3体系的还原性降低,Zr和Sn掺杂SrTiO3氧化还原性能提高,四种掺杂SrTiO3体系的带边位置仍然能够在水分裂过程中生成H2和O2.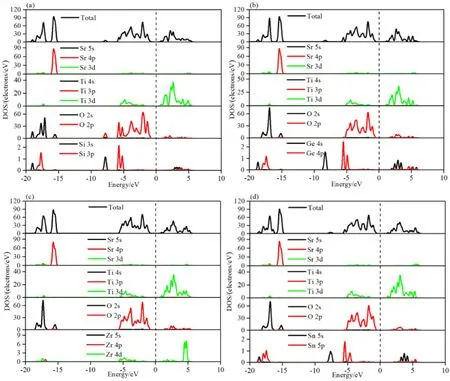
3.4 光催化性能分析

4 结 论
