汞在Ti2NO2 MXene表面吸附氧化的第一性原理计算
2024-01-18魏煜莹王军凯黄珍霞戚海新王向岭
魏煜莹,王军凯,黄珍霞,戚海新,王向岭
(1.河南理工大学 材料科学与工程学院,焦作 454003;2.河南理工大学 化学化工学院,焦作 454003)
1 引 言
汞(Hg)是一种有毒的重金属,一直被认为是“十大值得关注的化学物质”之一.自然界存在的汞主要由人为因素产生,如化石燃料燃烧、水泥生产、钢铁生产、有色金属冶炼、废物处理等都会造成汞的排放.其中,在人类生产生活中大量使用的化石燃料,其燃烧排放的汞含量占估计排放量的24%.元素汞(Hg0)具有高蒸汽压和低溶解度,因此难以从燃烧的烟气中去除[1].近年来,人为因素向自然界排放的汞已高于2000吨,汞通过食物链传递给人类,对人体造成了不可预估的伤害.Hg2+可损害肾脏和肺,甲基汞会成为一种神经毒素损害大脑功能[2-3].除此之外,由于汞的易挥发性和重金属的不可降解性,还会造成水和土壤的污染.
学者们为减少汞的排放进行了大量的研究[4],已经获得了一系列对Hg0脱除有前途的吸附剂和催化剂,包括活性炭[5]、金属氧化物[6-10]、金属硫化物[11,12]、h-BN[13]、纯金属[14-17]、聚合物[18,19]、水滑石[20]和金属有机构架[21]等.然而,由于成本等因素的限制,目前只有活性炭有望实现商业化应用,但是活性炭存在除Hg0效率一般且不可重复利用的缺点.虽然有研究[22]表明采用卤素改性活性炭能促进Hg0最终转化为卤化物,提高汞的去除效率,但该方法可能会造成二次污染.因此,进一步设计开发具有高效除Hg0特性的新材料具有十分重要的意义.
MXene是一种新型二维过渡金属碳化物/氮化物,自2011年首次报道以来[23],由于其优异的物理与化学性能,在材料领域逐渐成为研究热点[24].MXene的化学通式为Mn+1XnTx(n=1,2或3),其中M代表早期过渡金属,X是碳或者氮,Tx表示表面基团[25].由于MXenes通常是在水性介质中合成,因此其表面常含有-OH,-O,-F等基团[26].此外,理论计算表明,由-O基团修饰的MXenes通常更稳定.MXenes由于具有快速电荷转移、大表面积、亲水表面、可调带隙、高活性位点、表面基团丰富及低成本等优点,已被广泛应用于气体吸附、N2氨化、CO氧化、CO2还原等领域.目前,已有研究表明MXene材料也可用于吸附去除汞.Lee等人[27]通过水热法合成了高稳定性的磁性Ti3C2TxMXene纳米复合材料,并应用于水相吸附去除汞离子,结果表明,磁性Ti3C2TxMXene在广泛的pH条件下,可达1128.41mg/g的最大Hg离子吸收能力.在MXene的合成过程中,空缺通常是不可避免的.相关文献[28,29]表明单层MXene表面普遍存在不同的点缺陷,并且MXenes上存在的点缺陷或空位能够改善MXenes的性能.Gao等人[30]对汞在Ti2CO2(Mxenes)单层和具有一个氧空位缺陷Ov-Ti2NO2(MXenes)表面上的吸附氧化行为进行了研究,结果表明Hg0在Ti2CO2(MXenes)和Ov-Ti2NO2(MXenes)的吸附分别以物理吸附和化学吸附机制为主,并且有氧空位缺陷(Ov-Ti2NO2)的MXenes吸附氧化汞的性能比Ti2CO2(Mxenes)更优异.然而,目前MXenes吸附汞的研究主要集中在C系,关于汞在N系MXenes表面上的吸附行为和氧化机理尚未报道.
基于此,本文采用第一性原理计算的方法,研究了Hg0在Ti2NO2MXene和Ov-Ti2NO2MXene表面的吸附、氧化行为.相关研究结果对Hg0的吸附和氧化,减少汞的危害具有一定的理论指导和现实意义.
2 计算方法
采用Materials Studio软件中的Dmol3程序包进行第一性原理计算.所有计算均采用广义梯度近似(GGA)泛函PBE进行,布里渊区K点网格划分采用Monkhorst-Pack方法.在几何优化过程中,能量、力参数及最大位移的收敛标准分别为1×10-5Ha,0.002 Ha/Å,0.005nm.SCF计算收敛的电子密度变化小于1×10-6Ha.
选择3×3×1 Ti2NO2超胞(图1)作为衬底,计算采用3×3×1的K点网格.为了消除Z方向相邻盒子之间相互作用的影响,在该方向施加了30 Å的真空层.此外,从原始的3×3×1 Ti2NO2超胞中去除一个O原子形成表面带有氧空位的Ti2NO2,表示为Ov-Ti2NO2(图2).氧空位的形成能(Eform(Ov))按公式(1)计算:

图1 3×3×1Ti2NO2超胞俯视图和侧视图
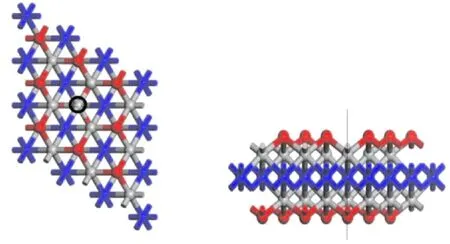
图2 优化后的Ov-Ti2NO2
Eform(Ov)= 1/2EO2+EOv-Ti2NO2-ETi2NO2
(1)
其中,1/2EO2、EOv-Ti2NO2和ETi2NO2分别描述了一半的O2分子、具有单个氧空位的Ti2NO2和原始Ti2NO2的优化后的总能量.
Hg0和HgO在Ti2NO2和Ov-Ti2NO2表面的吸附能(Eads)计算如下:
Eads=E(adsorbate/substrate)-Eadsorbate-Esubstrate
(2)
其中,E(adsorbate/substrate)、Eadsorbate、Esubstrate分别表示具有吸附Hg0或HgO的底物、气相Hg0或HgO和底物的总能量.根据吸附能的定义,能量越负,表明吸附作用越强.
Hg0在Ti2NO2和Ov-Ti2NO2表面氧化为HgO会涉及不同的反应态,包括初态(IS)、过渡态(TS)和最终态(FS).采用线性同步度越/二次同步度越(LST/QST)方法搜索Hg0氧化反应路径上的TS.TS与IS之间的能差称为Hg0氧化过程中的反应势垒.因此,反应势垒(Ebarrier)定义如下:
Ebarrier=ETS-EIS
(3)
3 结果与讨论
3.1 Hg0在Ti2NO2上的吸附
本文首先研究了Hg0在Ti2NO2上的吸附行为,考虑了Hg0在Ti2NO2上的四种不同吸附位点.图3(a)(b)(c)(d)为Hg0在Ti2NO2上四种可能的吸附位点:Hg0在Ti2NO2的O原子正上方(a),Hg0在Ti2NO2的Ti原子正上方(b),Hg0在Ti2NO2的N原子正上方(c),Hg0在Ti2NO2的O桥位中心上方(d).表1为Hg0在Ti2NO2上四种吸附构型的吸附能、几何参数以及Mulliken电荷[30].可以看出,第一种构型,优化后的Hg0在O原子正上方,这导致Hg0与Ti2NO2表面有较长的平衡距离(3.054 Å),该构型的吸附能为-33.70 kJ/mol.同时,约0.052e的电荷从Hg0转移到Ti2NO2.在Ti原子上方吸附的Hg0与Ti2NO2表面的距离为2.879 Å,该构型的吸附能为-36.08 kJ/mol,约0.061e的电荷从Hg0转移到Ti2NO2.在N原子顶部吸附的Hg0,与Ti2NO2表面之间的距离为3.020Å.吸附能为-36.73 kJ/mol,略大于其他三种构型.此外,Hg0向Ti2NO2提供了0.052e的电荷.Hg0在O桥位上方原子正上方,Hg0与Ti2NO2的距离为2.880Å,该构型的吸附能为-36.06 kJ/mol.同时,约0.061e的电荷从Hg0转移到Ti2NO2.
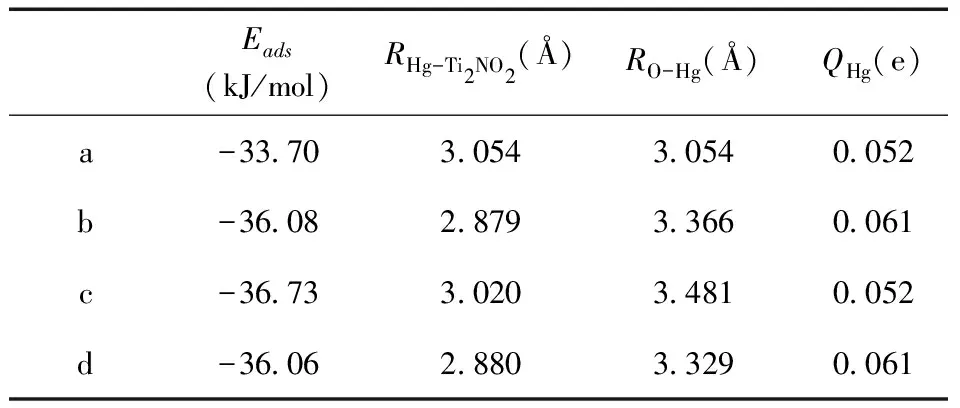
表1 Hg0在Ti2NO2上的吸附能、几何参数和密立根电荷
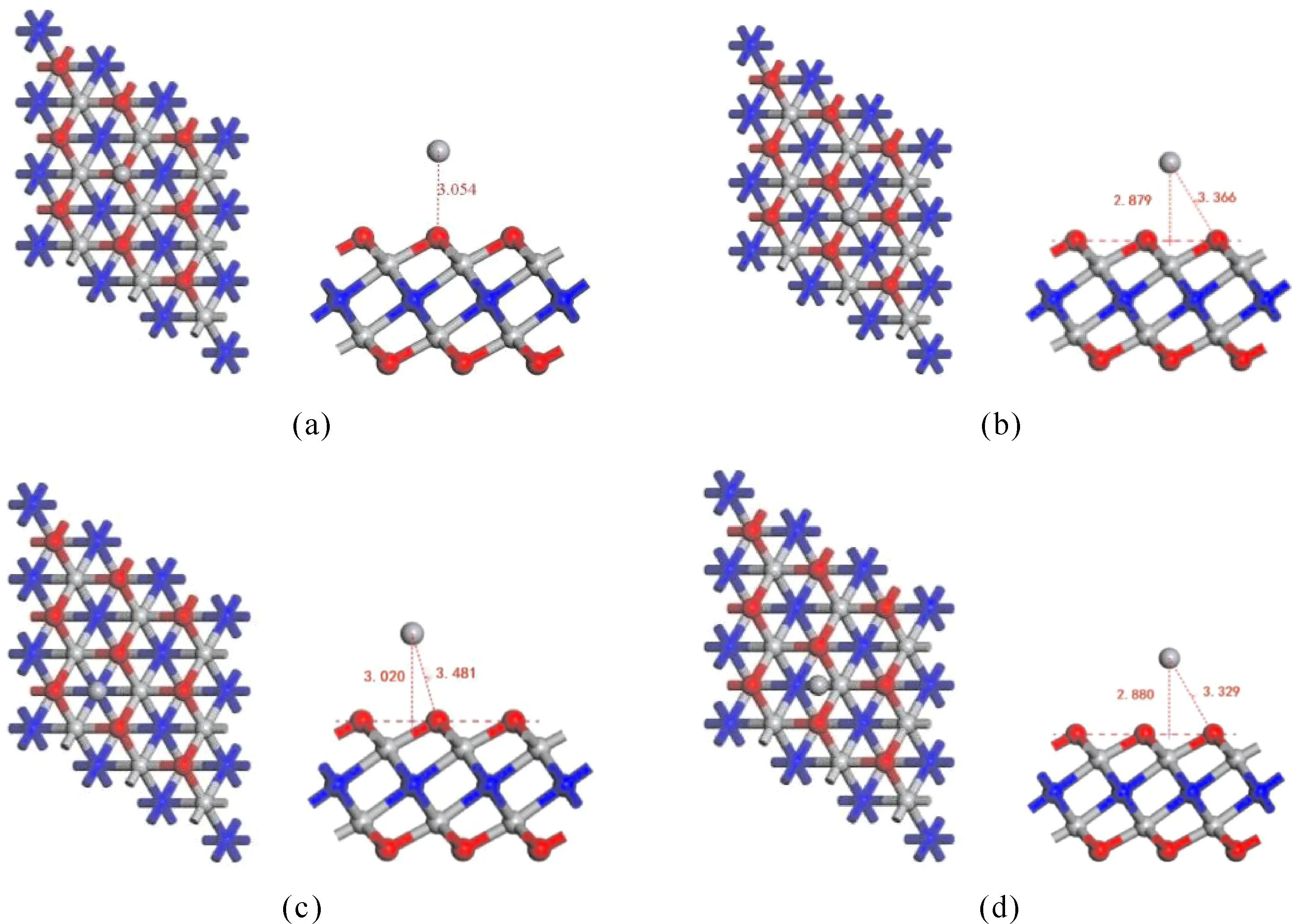
图3 Hg0在Ti2NO2上的不同位点的俯视图和侧视图:(a)O顶位,(b)Ti顶位,(c)N顶位,(d)O桥位
综上,Hg0更倾向于吸附于N原子顶端,Hg0在Ti2NO2上的吸附属于物理吸附.
3.2 Hg0在Ov-Ti2NO2上的吸附
进一步研究了Hg0在Ov-Ti2NO2上的吸附行为.Hg0在Ov-Ti2NO2表面的吸附只有一种稳定的吸附构型,即Hg0在O空位顶部的吸附.优化后的结构中(图4),Hg0距离相邻的O原子3.386 Å,距离Ov-Ti2NO21.591 Å,约为Hg0距离Ti2NO2的一半.Hg0在Ov-Ti2NO2上的吸附能为-104.84 kJ/mol,远远大于在Ti2NO2上的吸附能.同时,约0.226e的电荷从吸附的Hg0转移到Ov-Ti2NO2上.以上结果均表明,Ov-Ti2NO2对Hg0吸附能力远高于Ti2NO2.
图5显示了Hg0在Ov-Ti2NO2表面吸附前后的体系中Hg、O、Ti和N原子的分波态密度(PDOS).如图5(a)和(c)所示,与Ov-Ti2NO2吸附Hg0前相比,Hg0吸附在Ov-Ti2NO2后,所有轨道都明显向较低的能级移动.而Ov-Ti2NO2吸附Hg0前后表面O、Ti和N原子的轨道吸附前后没有明显的变化,这是因为O、Ti、C原子的直径比Hg原子的直径要小,因此O、Ti和N原子的电子轨道很难被极化.Hg0被吸附后,Hg的d轨道在值为-6.61 eV处与Ti原子的s轨道和d轨道杂化.Hg的s轨道也在-1.84 eV和-4.24 eV与O和N原子的p轨道和Ti原子的d轨道重叠.综上所述,在气体Hg0和Ov-Ti2NO2之间轨道杂化和重叠可能会带来显著的相互作用,从而增强了Hg0在Ov-Ti2NO2上的吸附.
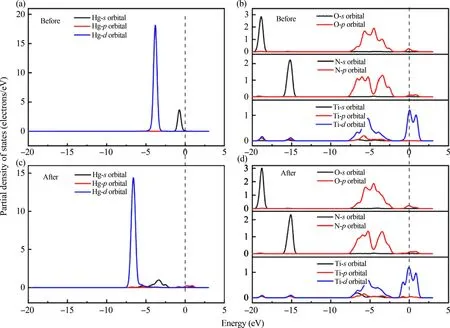
图5 Hg0在Ov-Ti2NO2上吸附前后的分波态密度
3.3 HgO在Ti2NO2和Ov-Ti2NO2上的吸附
Hg0在Ti2NO2和Ov-Ti2NO2表面最终会氧化生成HgO,研究HgO与Ti2NO2和Ov-Ti2NO2之间的相互作用对理解Hg0转化HgO过程起着至关重要的作用.因此,进一步研究了HgO在Ti2NO2和Ov-Ti2NO2表面的吸附行为.考虑了HgO可能的吸附取向和可能的吸附位点.优化后得到的HgO在 Ti2NO2表面的吸附结构及其吸附能以及几何参数分别如图6和表2所示.研究发现,图6(a)构型最稳定,其吸附能为-110.49 kJ/mol.此时,O原子在上,Hg原子在下,二者形成的键长为2.1 Å.HgO被吸附在Ti2NO2表面,并与表面的O原子形成化学键,键长为2.5 Å.图6(b)构型吸附能为-151.46 kJ/mol,数值上大于图6(a)构型的吸附能,但是O原子指向表面的Ti原子,并与其形成化学键,导致Hg-O键断裂,形成Ti-O键.图6(c)构型中,HgO在N原子的正上方,与临近的O原子距离2.1Å,与图6(a)和(b)的构型相比,它的吸附能较低(-71.42 kJ/mol).根据以上结果可知,HgO在Ti2NO2上的吸附为化学吸附.
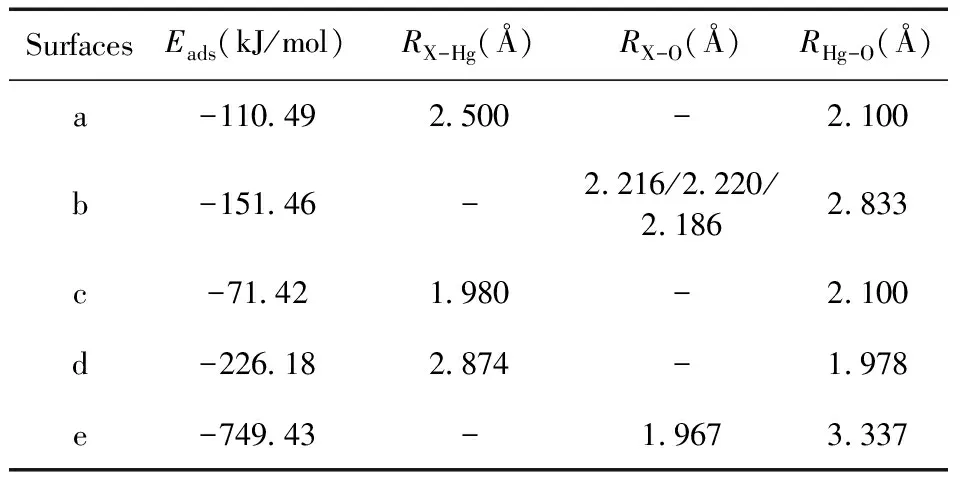
表2 HgO在Ti2NO2上的吸附能、几何参数,X表示表面T或O原子
HgO在Ov-Ti2NO2上的吸附如图6(d)和6(e)所示.在图6(d)构型中,HgO垂直吸附在Ov-Ti2NO2表面并形成三个Hg-Ti 键,吸附能为-226.18kJ/mol.吸附后HgO中Hg-O键长和形成的Hg-Ti的键长分别为1.978 Å和2.874Å.图6(e)构型的吸附能更高(-749.43 kJ/mol),但是在图6(e)构型中发现,HgO在Ov-Ti2NO2上吸附后,HgO的Hg-O键完全断裂,Ov-Ti2NO2上的O空位由产生的O原子填充.这些结果表明,HgO在Ov-Ti2NO2表面的吸附也为化学吸附,且吸附强度强于在Ti2NO2表面的吸附.
3.4 Hg0转化为HgO的反应过程
以往的研究表明,Hg0可以被催化剂/吸附剂的表面氧氧化.催化剂/吸附剂表面的氧在Hg0的氧化反应中起着重要的作用.在Hg0的氧化过程中,Hg0首先吸附在Ti2NO2或Ov-Ti2NO2上,然后与氧官能团反应形成HgO.而后在Ti2NO2或Ov-Ti2NO2表面解吸形成游离HgO.为了深入了解Hg0在Ti2NO2和Ov-Ti2NO2表面的氧化过程,计算了氧化反应的能垒.研究了含氧官能团的Ti2NO2和Ov-Ti2NO2将Hg0转化为HgO的过程,并根据Hg0和HgO在Ti2NO2和Ov-Ti2NO2上的吸附结构,确定了反应途径中涉及的初态(IS)、过渡态(TS)和最终态(FS)构型.
图7显示了Hg0在Ti2NO2表面转化的能量分布,以及相应的初态(IS1)、过渡态(TS1)和最终态(FS1)的优化构型.Hg0首先吸附在Ti2NO2表面O位点上形成IS1,而后表面的O原子从Ti位迁移到吸附的Hg位置,并与吸附的Hg反应生成HgO,此为FS1.该反应的能垒为101.42 kJ/mol.同时发现,FS1的总能量高于IS1,说明这一步是一个吸热反应.此外,在解吸步骤中,需要约110.49 kJ/mol的外部能量才能形成游离HgO,该过程也是吸热的.
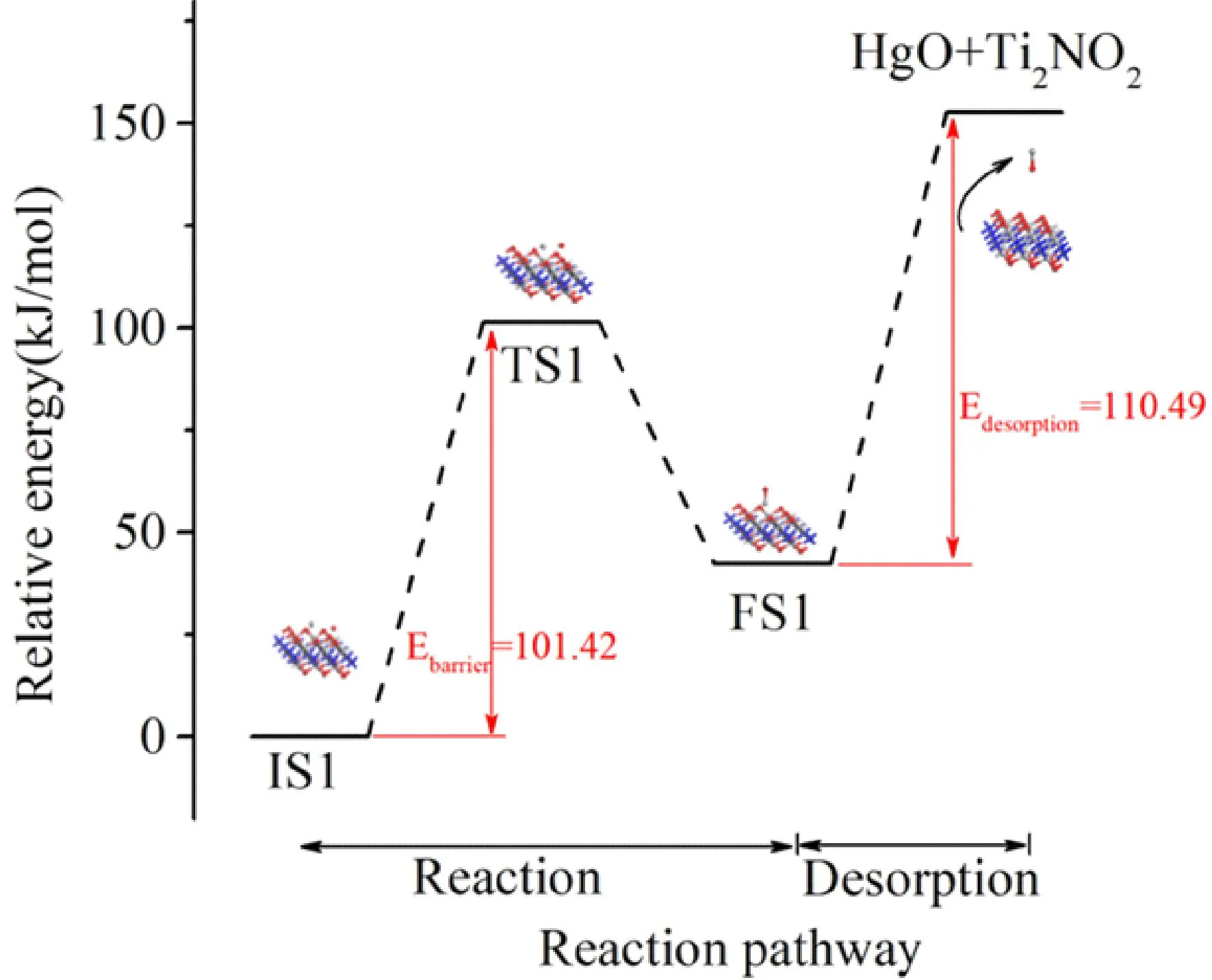
图7 在Ti2NO2上的非均相Hg0转化的能量分布和优化构型
Hg0在Ov-Ti2NO2上转化的能量分布和优化构型如图8所示.Hg0首先被化学吸附在Ov-Ti2NO2上形成初态(IS2).而后,O-O键断开,自由O原子向被吸附的Hg原子移动,同时与Hg原子发生反应生成HgO,此为最终态(FS2).该氧化反应是一个能垒为92.55 kJ/mol的放热过程.在解吸步骤中,形成的HgO分子从Ov-Ti2NO2表面脱附需要226.18 kJ/mol的外部能量.
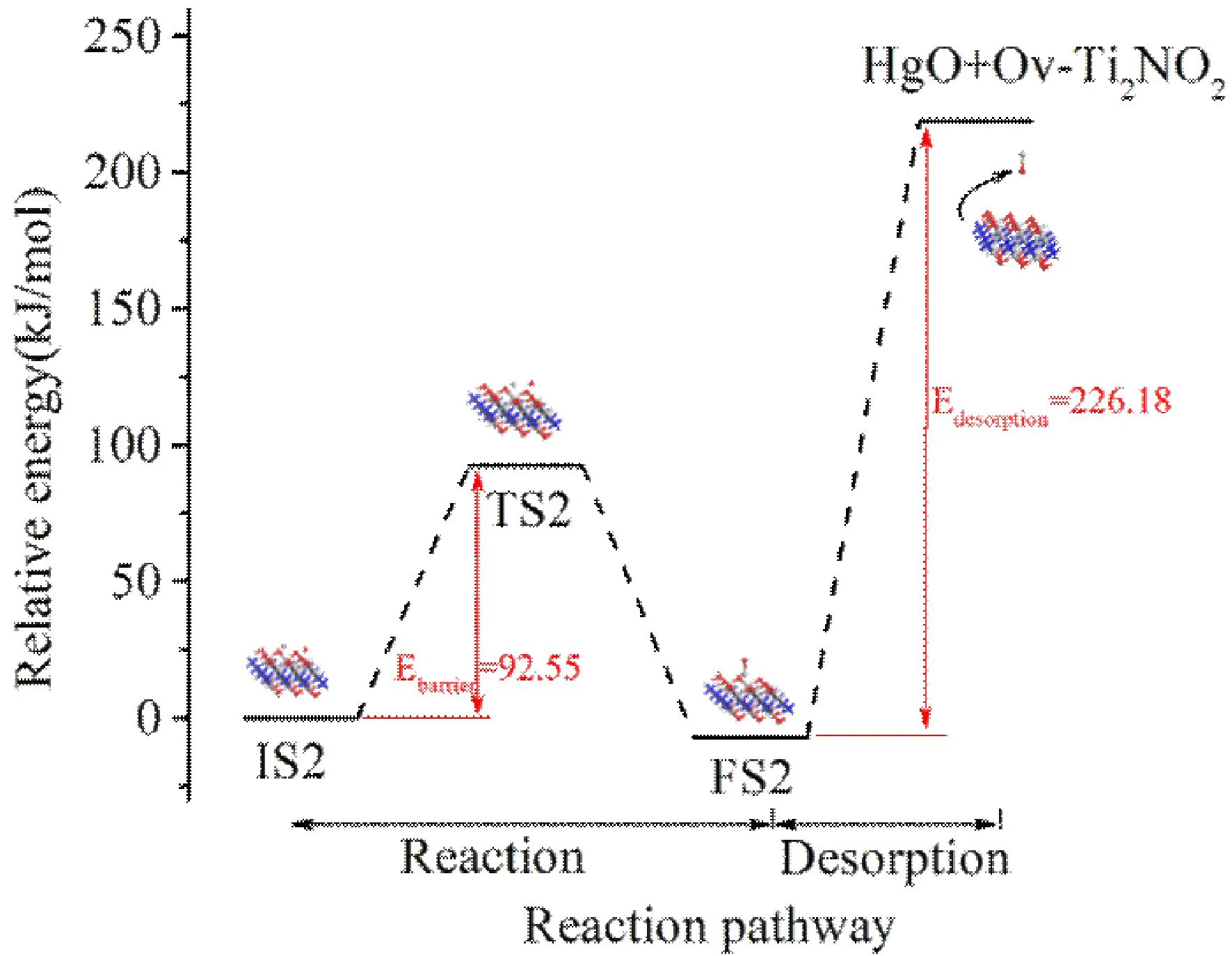
图8 在Ov-Ti2NO2上的非均相 Hg0转化的能量分布和优化构型
从以上结果可以看出,Hg0在Ti2NO2表面氧化的能垒(101.42 kJ/mol)高于在Ov-Ti2NO2表面(92.55 kJ/mol),表明Ov-Ti2NO2比Ti2NO2更有利于Hg0的氧化.进一步对比了Hg在其他材料表面氧化的能垒,结果发现:Hg0在V2O5/TiO2(0 0 1)表面氧化的能垒为143.9~151.2 kJ/mol[31],在ZnSe(110)表面氧化的能垒为185.09 kJ/mol[32],均远高于Hg0在Ov-Ti2NO2表面氧化为HgO的能垒(92.55 kJ/mol).表明Hg0在Ov-Ti2NO2表面更容易发生氧化反应,Ov-Ti2NO2有望成为高效的除Hg0材料.此外,与Ti2NO2相比,生成的HgO从Ov-Ti2NO2表面脱附需要相对较高的外部能量(226.18 kJ/mol),表明氧化生成的HgO可以稳定吸附在Ov-Ti2NO2上,难以发生解吸导致二次汞污染.这非常有利于对汞的集中控制.
4 结 论
本文采用第一性原理计算分别研究了Hg0在Ti2NO2MXene和Ov-Ti2NO2MXene表面的吸附、氧化行为.主要结论如下:
(1)Hg0在Ti2NO2上的吸附较弱,而在Ov-Ti2NO2表面Ti原子上方的吸附较强.
(2)Hg0在Ov-Ti2NO2表面转化为HgO的能垒为92.55 kJ/mol,低于Ti2NO2表面的101.42 kJ/mol,表明Hg0在Ov-Ti2NO2表面更容易发生氧化反应.
(3)产物HgO在Ov-Ti2NO2表面脱附需要226.18 kJ/mol能量,远高于在Ti2NO2表面脱附所的110.49 kJ/mol,说明Ov-Ti2NO2对产物HgO的集中控制能力优于Ti2NO2,从而更能抑制HgO脱附造成二次污染.
