密度泛函理论研究KA油在氧化钨表面上的吸附分离
2024-01-18庞浩成容铠东方岩雄
庞浩成,容铠东,何 东,李 旭,方岩雄
(广东工业大学 轻工化工学院,广州510006)
1 引 言
环己烷(Cy)氧化生成环己醇(CyOH)和环己酮(Cy=O)是重要的化工反应,该反应的混合产物被称为KA油.其中,环己酮作为化工生产中重要的中间体,广泛的用于生产尼龙-6[1-4].在工业生产中,由于环己醇和环己酮的沸点较为接近[5],采用真空精馏的方式分离二者.然而,该过程能耗大,且需大量水资源,对环境和经济方面造成严重负担.此外,在分离过程中环己醇和环己酮会形成竞争吸附[6],致使分离效率较低.因此,开发设计有效分离KA油的新材料,提高工业分离效率和经济效益,这些重要的科学问题受到越来越多的研究工作者广泛关注.
氧化钨(WO3)作为一种具有独特物理化学性质的半导体功能材料,广泛的应用于催化、环境、能源、信息科学等领域[7-9].由于氧化钨具有较窄的禁带宽度(Eg = 2.5~2.8 eV),可吸收480 nm以下的光,能诱导光子的直接跃迁,因此在光催化氧化反应中有着重要应用[10].文献报道,Zheng等[11]制备了一种Au/WO3复合的催化剂,使环己烷氧化转化效率提高到商业化的两倍.Xiao等[12]制备了Ag纳米粒子负载的单层WO3纳米片材料,在室温光照下可有效催化环己烷氧化生成KA油.该研究使用的氧化钨为六方晶系(h-WO3),其表面的O原子缺陷和不饱和W原子,可有效的活化、吸附客体分子.Ueyama等[13]通过同位素标记实验和ESR测试证明,环己自由基的存在,其与WO3晶格中缺陷O原子作用,致使环己酮的形成.
密度泛函理论(DFT)是基于多电子体系电子结构的研究方法,广泛用于研究材料的吸附,催化,反应机理,分子的结构与性质,是计算化学与实验数据相结合最重要的技术手段[14,15].采用DFT方法,研究WO3材料的制备工艺、性能和应用具有重要意义.Cao等[16]报道了Pd修饰的h-WO3,用于丙醇气体敏感机制的研究.Nagarajan[17]和Zhao[18]等研究发现h-WO3表面的O原子与CO气体分子存在主-客体电子转移现象.Li等[19]基于DFT方法,研究HN3、NO、H2S和HCHO气体在WO3表面的吸附,通过表征部分态密度(PDOS)、带隙和功函数揭示了WO3在传感器领域的中的潜应用.Hurtado-Aular等[20]采用DFT发方法研究了CO2和H2O在W3Ox/M(111)材料(x = 6和9,M = Cu、Ag和Au)上的吸附,计算结果表明氧化物的化学计量决定了电荷转移的方向,W3Ox的表面在吸附后发生氧化/还原反应.Tian等[21]构建了不同的h-WO3(001)表面O终端和WO终端模型,DFT计算结果显示,O终端模型具有较低的表面能,且最稳定、存在可能性更高,此外正是由于这种O终端的存在,才使h-WO3吸附CO中表现出优异性能.Albanese等[22]采用DFT计算发现H2O更喜欢吸附在h-WO3表面的不饱和W原子上,这是由于离子型氧化物的不饱和W原子可视为路易斯酸位.Qin等[23]研究发现,NO2分子可以以多种稳定构象吸附在WO3表面上,从而产生新的电子组态,改变了带隙和费米级.
因此,本文基于DFT理论计算,构建了h-WO3(001)理论模型,用于研究KA油在干净和有机物修饰的h-WO3(001)表面上的吸附能和吸附构型;研究不同的吸附位点和吸附构型对吸附的影响;计算态密度(DOS)、部分态密度(PDOS)和差分电荷密度(CDD),表征吸附过程中的主-客体电荷转移方向和电子轨道杂化;采用憎水剂六甲基二硅醚和硅烷[24-26]作为修饰基团对h-WO3(001)表面改性,探究其在分离环己酮和环己醇的中的潜在应用.
2 理论模型和方法
本研究工作采用六方晶系的h-WO3构建理论模型,其空间群为P63cm(a=b= 7.32 Å,c= 7.66 Å,α=β= 90°,γ= 120°)[27].在原始的h-WO3的晶胞中存在两种结晶学不等价的O原子,故产生两种W-O键长,分别为2.00 Å和1.88 Å(图1a).随后,构建了沿(001)晶面具有O终端[21]和缺陷O的2×2的超晶胞(图1b).该模型包含3个原子层,总计原子数为160,沿z方向的真空层厚为15 Å,以避免层间相互作用.图1b中的符号V代表O缺陷的位置.在计算过程中,顶层原子保持弛豫,其他原子固定其坐标位置.本研究考虑四种可能的吸附位点(图1c).A:五配位钨原子(W5);B:六配位钨原子(W6);C:与两个六配位钨原子连接的氧原子(W6-O-W6);D:与六配位钨原子和五配位钨原子连接的氧原子(W6-O-W5).基于客体分子Cy、CyOH和Cy=O在每个吸附位点上考虑两种吸附取向,即垂直(V)和平行(P).客体分子吸附在h-WO3(001)表面时,其构型表示为X-Y(X=A、B、C、D,Y=P、V).

图1 (a)氧化钨晶胞模型,(b)具有氧缺陷的氧化钨超晶胞模型,(c)考虑的四种吸附位点(O,红;W,蓝)
本研究工作采用的DFT计算在Materials Studio 软件中的Dmol3[28,29]模块完成.模型的几何优化基于广义梯度交互泛函GGA/PBE[30-32].采用Double Numerical plus d-functions (DND)基组和DFT Semi-Core Pseudopots (DSPP)赝势处理.结构优化的能量、力和位移的收敛标准分别为2.0×10-5Ha、4.0×10-3Ha/Å和5.0×10-3Å.基于CASTEP[33,34]模块计算差分电荷密度(CDD)表征主-客体电荷转移情况.为表征电子结构,进行了态密度(DOS)计算.为了选择合适的泛函来描述h-WO3(001)模型,分别使用PBE、PW91和PW91-OBS泛函来计算晶胞的参数和W-O键长.计算结果列于表1,与实验数据比较发现,三种泛函计算得到的键长无明显差异,而PBE泛函广泛应用于氧化钨的DFT研究中[35-37],因此本文选择PBE泛函进行计算.吸附能(Eads)计算公式如下所示:
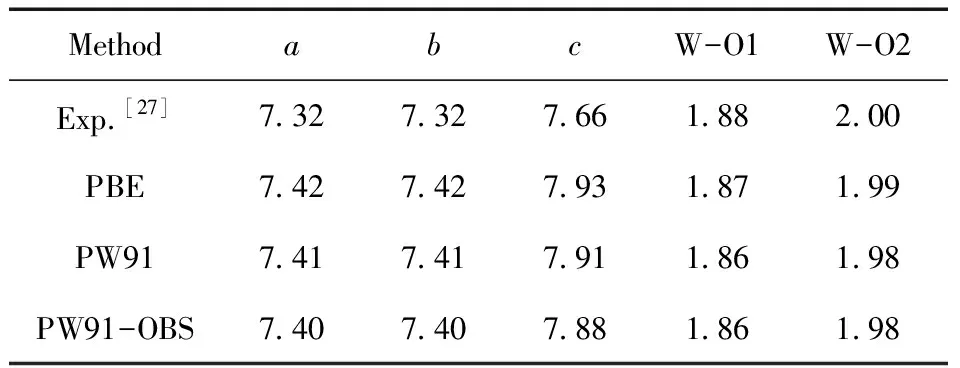
表1 h-WO3晶格常数和W-O键长(Å)
Eads=Etot-Eguest-Esur
(1)
Etot系统的总能量,Eguest客体分子的能量,Esurh-WO3(001)表面的能量.计算的吸附能是吸附物和底物的电子几何特性的函数[38],表征主-客体分子之间作用的强弱.
3 结果与讨论
3.1 吸附在干净的h-WO3(001)
Cy吸附在h-WO3(001)上的吸附能列于表2中,吸附构象和最近的作用距离置于图2中.从图2中可以看出,Cy以垂直吸附构象在W5原子上(A-V型)具有较大的吸附能-44.4 kJ·mol-1,最近作用距离为2.10Å.
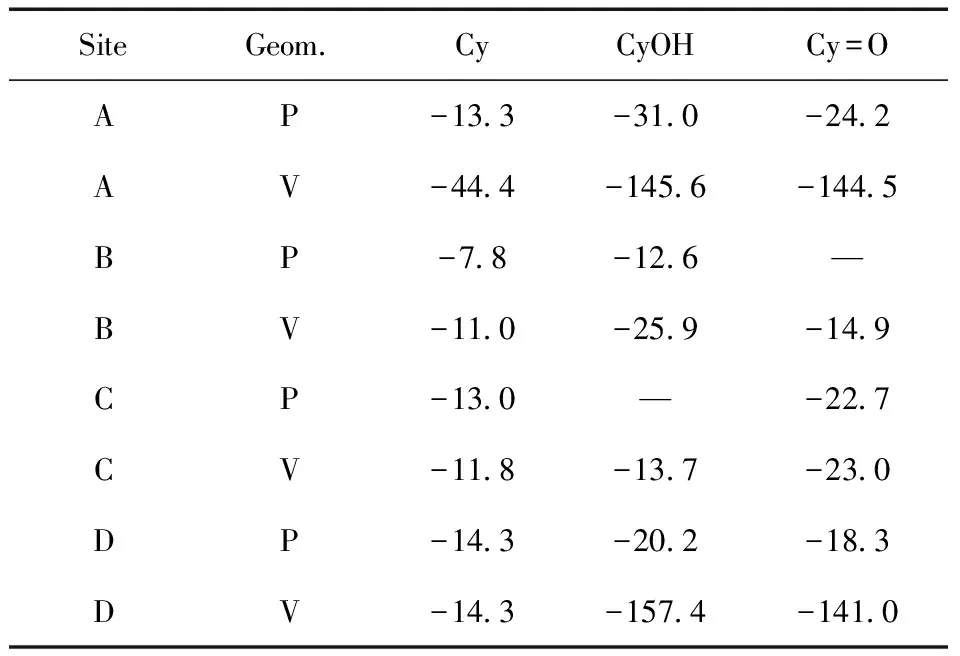
表2 客体分子在干净的h-WO3(001)吸附能(kJ·mol-1)
而CyOH偏好于垂直吸附在W6与W5原子之间的桥O上(D-V型),其吸附能为-157.4 kJ·mol-1,最近W5…OCyOH作用距离为2.15 Å;Cy=O与Cy的吸附构象相类似,以垂直吸附构象在W5原子上(A-V型)吸附能为-144.5 kJ·mol-1,最近W5…OCy=O作用距离为2.12 Å.此外,CyOH在三者中具有最大的吸附能是由于其OH基团与表面的桥O形成了氢键,即HCyOH…OWO3的距离为1.85 Å.在几优化过程中,Cy=O与CyOH分别在B-P型和C-P型未获得稳定构象.
为研究客体分子与h-WO3(001)表面的电荷转移,我们以客体分子最优的吸附构象计算并绘制了其吸附前后的差分电荷密度图(图2d~f).图中蓝色区域表示电荷的增加,黄色区域表示电荷减少.我们发现客体分子均与h-WO3(001)表面有电荷转移现象.W5周围的电子云呈现黄色即为电子从W原子上减少,而Cy=O与CyOH分子中(图2e-f),其O原子周围电子云呈现蓝色,即在吸附之后电子有所增加,因此电荷转移的方向是从h-WO3(001)的W5处到客体分子上.此外,与Cy吸附不同的是,CyOH和Cy=O吸附时的差分电荷密度图中,蓝色区域与黄色区域直接接触,且电子云面积更大,说明相互作用更强烈.如果用电负性描述分子中原子吸引电子的倾向,O原子的电负性是18.6 eV·e-1,比C原子(13.9 eV·e-1)和H原子(13.6 eV·e-1)大[39],与我们计算的结果相一致,即CyOH和Cy=O官能团的O原子,比Cy中的C原子和H原子周围蓝色电子区域面积大,说明O原子上积累的电荷更多.同时,CyOH和Cy=O的吸附能比Cy更负,对应吸附作用更强.
为了进一步阐释客体分子和h-WO3(001)的电子轨道杂化情况,计算了客体分子最佳吸附构象的部分状态密度(PDOS).如图2(g)所示,Cy中的H原子的2s轨道和h-WO3(001)表面的W5原子的5d轨道的PDOS没有太多的重叠,说明其吸附主要是物理吸附.图2(h)显示,CyOH中的OH基团的O原子的2p轨道与表面的W5原子的5d轨道在-9.0 eV、-7.4 eV和-4.6 eV处重叠;图2(i)显示,Cy=O的羰基O原子的2p轨道与表面W5原子的5d在-9.0 eV、-7.7 eV和-6.5 eV处重叠.我们可以发现,CyOH和Cy=O的吸附主要是源于官能团中的O原子的2p轨道和h-WO3(001)表面的W5原子的5d轨道之间的强轨道杂化[40],是化学吸附.综上所述,我们发现,在干净的h-WO3(001)表面上,客体均可以吸附,但Cy是理吸附,CyOH和Cy=O是化学吸附存在电子转移,轨道杂化的现象.两者的吸附能较为相近在分离过程中会存在竞争吸附,难于分离,该计算结果与实验观察一致.
3.2 六甲基二硅醚修饰的h-WO3(001)
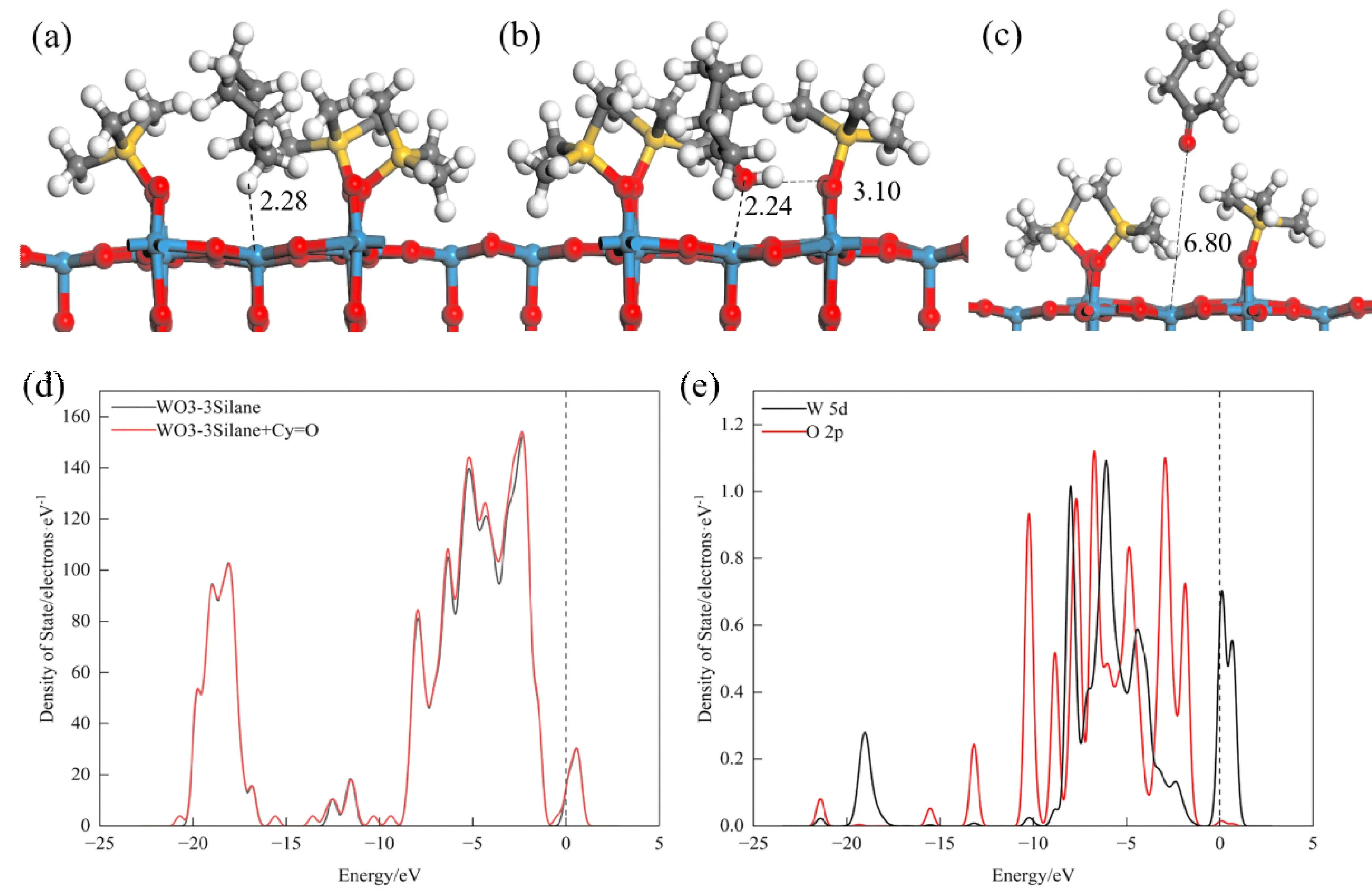
我们尝试用六甲基二硅醚(HMDSO)修饰h-WO3(001)表面,计算客体分子在表面的吸附能以解决CyOH和Cy=O难分离的问题.我们建立了不同数量HMDSO修饰WO3(001)表面的模型,客体分子的吸附能列于表3.最佳吸附构象,差分密度图和部分态密度图均在图3中给出.从表3可以看出,客体分子在一个HMDSO基团修饰的WO3(001)上具有最大的吸附能,其值递变顺序为Cy (-21.3 kJ·mol-1) 图3 在HMDSO修饰的h-WO3(001)上的最优吸附构象和最近作用距离(Å):(a)环己烷,(b)环己醇,(c)环己酮;差分电荷密度图:(d)环己烷,(e)环己醇,(f)环己酮;(g)修饰前后表面态密度图;(h)环己醇和(i)环己酮吸附的部分态密度图(O,红;W,蓝;C,灰;H,白;Si,黄) 图3(a~c)给出了客体分子在HMDSO修饰的h-WO3(001)表面的吸附构型,Cy,CyOH和Cy=O与表面的W5原子的最近距离分别为3.10 Å、2.22 Å和2.15 Å.该距离明显比在干净的WO3(001)上(2.10~2.15 Å)长.这是由于HMDSO基团的空间位阻作用,使客体分子被推向远离活性W5原子处.图3(d~f)的差分密度图显示,CyOH和Cy=O与表面的W5原子有明显的电荷转移现象.此外,通过比较干净的与HMDSO修饰的WO3(001)表面的态密度,图3(g)所示,我们发现修饰后整体态密度向左移动,说明体系的能量更低,意味着修饰的表面更稳定[41],与我们计算的客体分子的吸附能更低的结论相吻合.图3(h)中,CyOH的O原子的2p轨道与W5原子的5d在大约-10.0 eV、-8.0 eV、-6.0 eV、-4.3 eV处重叠,表明CyOH的O原子与W5原子的轨道杂化,验证其为化学吸附,结合差分密度图我们可以知道,CyOH吸附之后从表面获得电子.相似的情况也发生在Cy=O的吸附过程,从图3(i)中可以看出Cy=O的O原子的2p轨道与W5原子的5d轨道在-10.0 eV和-9.0 eV处有重叠,电子轨道的高度杂化[42],即为化学吸附;吸附之后,导带也是由O原子的2p轨道提供,表面电子聚集在Cy=O的O原子上.综上,在HMDSO修饰的h-WO3(001)上Cy的吸附远小于0.8 eV属于物理吸附[42,43].而CyOH和Cy=O的吸附较大,且存在电子转移的现象为化学吸附,同时二者的吸附能差43.96 kJ·mol-1,足以通过吸附将其分离. 本研究工作中,我们还尝试用三甲基硅烷(TMS)修饰h-WO3(001)表面,由于TMS基团空间位阻较小,我们考虑不同量对客体分子吸附能的影响,计算获得的吸附能列于表3.最佳吸附构象,差分密度图和部分态密度图均在图4中给出. 图4 在3个TMS修饰的h-WO3(001)上的最优吸附构象和最近作用距离(Å):(a)环己烷,(b)环己醇,(c)环己酮;(d)环己酮吸附前后的态密度图,(e)环己醇吸附后的部分态密度图(O,红;W,蓝;C,灰;H,白;Si,黄) 从表3中我们可以发现,Cy在两个TMS修饰的h-WO3(001)表面具有就最大吸附能-40.9 kJ·mol-1,其值仍小于0.8 eV,仍为物力吸附.相类似的Cy=O在修饰TMS数量为2时,吸附能最大-131.1 kJ·mol-1,为化学吸附;当修饰量为3时,吸附能降为-29.8 kJ·mol-1,为物理吸附.TMS的修饰数量小于4时,CyOH仍具有较高的吸附能(-128.5,-132.4和-136.9 kJ·mol-1).该计算结果表明,当修饰TMS数量为3时,CyOH和Cy=O的吸附能相差102.6 kJ·mol-1,由此可以对二者进行分离.该现象可以从图4中得以验证,TMS修饰量为3时,客体分子的吸附构象,Cy和CyOH和Cy=O与表面的W5原子的距离分别为2.28 Å,2.24 Å和6.80 Å.此时,Cy=O被TMS修饰基团排斥远离表面,而对CyOH吸附具有选择性.从图4(d)可以看出,给Cy=O吸附前后的部分态密度几乎没有变化,即无轨道杂化,表面与Cy=O无键合[44],表现为物理吸附,吸附能较小.图4(e)显示出CyOH吸附在TMS修饰时,存在其羟基的O原子2p轨道和W5原子5d轨道在在-1.0 eV和-9.0 eV区域之间的重叠,表明二者之间的轨道杂化现象.此外,费米级以下的价带主要由羟基O原子的2p轨道提供,表明电荷主要聚集在O原子周围.总之,可以通过修饰3个TMS在h-WO3(001)上,调节吸附能的差异来有效分离CyOH和Cy=O.该理论结果为实验提供了可行性依据. 本研究工作,通过密度泛函理论计算了Cy,CyOH和Cy=O作为KA油混合物的客体分子,在干净的和HMDSO,TMS修饰的h-WO3(001)表面上的吸附能,吸附构型,差分密度和部分态密度.计算结果表明,Cy的吸附较小,表现为物理吸附.CyOH和Cy=O分子在干净的表面上吸附能较大,为化学吸附.此外,吸附能的大小受吸附位点和构象的取向性影响,其中在垂直吸附构象中,CyOH和Cy=O与表面的不饱和路易斯位点W5原子存在最近作用距离,具有明显的电荷转移现象.调节不同有机官能团修饰的h-WO3(001)表面,可以将CyOH和Cy=O有效的分离.采用HMDSO修饰时,表面对Cy=O具有选择性,表现出最大的吸附能,并伴随着电荷转移现象;TMS修饰,且数量为3时,表面对CyOH具有选择性,吸附能最大,为化学吸附;对Cy=O表现出排斥,吸附能较小,物理吸附的行为.该理论计算结果为实验分离KA油混合物提供了理论依据,加快了定向设计、合成、修饰功能材料的发展.此外,理论工作先于实验的研究思路,可以缩短材料的研发周期,节省人力,物力,资源,实现提高经济效益的目的.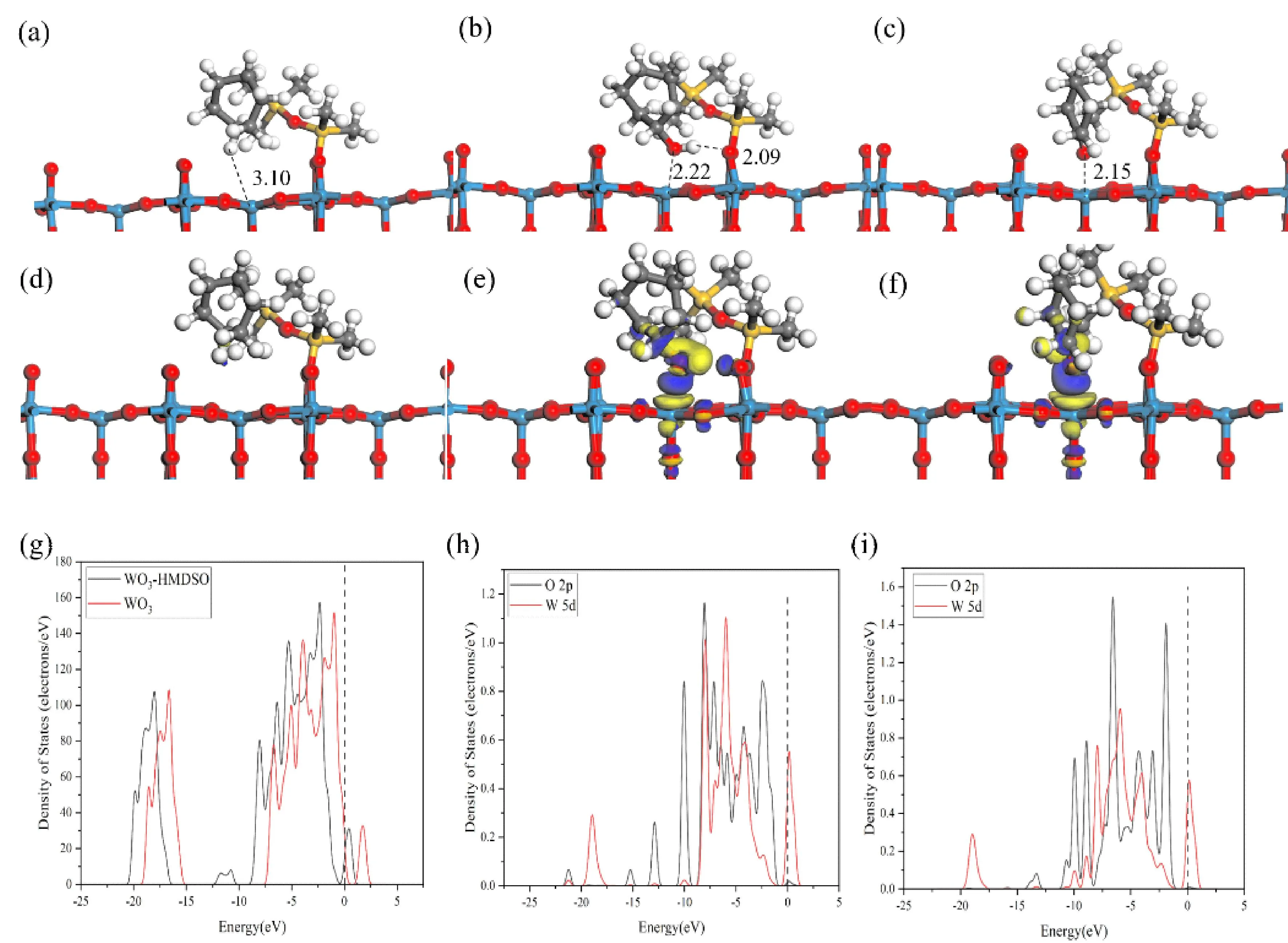
3.3 三甲基硅烷修饰的h-WO3(001)
4 结 论
