Fe掺杂碳纳米管吸附甲硝唑的第一性原理计算
2024-01-18李凤凤王军凯黄珍霞王一菲魏煜莹蔡艺璇
李凤凤,王军凯,黄珍霞,王一菲,魏煜莹,蔡艺璇
(1.河南理工大学 化学化工学院,焦作 454003; 2.河南理工大学 材料科学与工程学院,焦作 454003)
1 引 言
甲硝唑(MNZ)是一种抗生素和抗原虫剂,主要用于治疗或预防厌氧菌引起的系统或局部感染[1],如消化道、腹腔[2]和女性生殖系[3]等产生的厌氧菌感染,还能有效治疗乳腺导管扩张[4]和恶性伤口恶臭[5]等.然而,由于MNZ疗效高且成本低,常被一些国家违禁用于家禽,猪和鱼类以消灭寄生虫[6-9].这种抗生素难以生物降解且水溶性高,去除MNZ在技术上也具有很强的挑战性[10-12],从而导致了水溶液,土壤和粮食的污染,被人类误用后会发生一定程度的抗生素中毒,长时间饮用含抗生素的水源,不仅危害肠道,还会降低人体免疫力,从而影响人类健康[13].因此,研究除去水中的MNZ势在必行.
碳纳米管(CNT)是一种由六边形排列的碳原子构成数层到数十层的同轴圆管.自1991首次发现[14]以来,以其优异的性能引起了学者们的广泛关注[15-17].由于CNT具有比表面积大等特点,已有许多研究人员尝试采用CNT作为吸附剂[18-25].Larciprete等[26]通过密度泛函理论对比研究了原始CNT和Rh-CNT对NO2分子的吸附行为.研究表明:在200 K时,CNT上没有NO2吸附,但在CNT上掺杂Rh原子后,NO2与Rh产生了强烈的相互作用.Yoosefian[27]利用第一性原理计算研究了烟草烟雾中的亚硝胺在单壁CNT和负载Pd和Ni的单壁CNT上的吸附行为.结果表明:单壁CNT吸附亚硝胺分子的吸附强度一般,而负载Pd和Ni的单壁CNT,对亚硝胺分子表现出很强的亲和力,吸附能力显著增强.Zhang等[28]以CNT为原料,将漆酶(Laccase)直接固定在CNT上得到Laccase-CNT,分别用CNT和Laccase-CNT去吸附废水中的染料分子.结果表明:与原始CNT相比,Laccase-CNT对废水中的染料分子吸附能更大,电荷转移更多,去除率更高.Demir和Fellah[29]采用密度泛函理论研究了(4,0)单壁CNT和Pt掺杂(4,0)单壁CNT去对CO分子的吸附行为.研究表明:CO分子不能被吸附到固有的CNT上,而用Pt掺杂(4,0)CNT对CO却具有优异的吸附效果.这些研究都说明了掺杂原子后的CNT的性质发生改变,且吸附能力显著增强.An[30]等采用第一性原理方法研究了SO2分子在原始CNT和Fe-CNT上的吸附行为.结果表明:SO2分子在原始CNT上是弱物理吸附,而掺杂Fe后,形成了非常稳定的化学吸附.此外,Zhang等[31]采用密度泛函理论研究了空位缺陷(8,0)单壁CNT和Fe掺杂空位缺陷(8,0)单壁CNT对SO2分子的吸附行为.研究表明:Fe掺杂空位缺陷(8,0)单壁CNT对SO2的吸附能远高于空位缺陷(8,0)单壁CNT.以上研究结果表明Fe-CNT是一种优异的吸附材料,然而目前尚缺乏Fe-CNT对MNZ吸附行为的研究.
基于此,本文采用第一性原理的计算方法,研究了Fe-CNT对MNZ的吸附行为.相关研究结果对MNZ的吸附,减少其危害具有一定的理论指导和现实意义.
2 计算方法
采用Materials Studio构建(5,5)CNT初始模型,末端碳原子被氢原子饱和,以保持电荷中性,避免悬垂键,减少边缘效应.其结构有100个碳原子和20个氢原子,直径和长度分别为6.78 Å和12.30 Å.采用DMol3模块实现第一性原理计算.交换相关泛函和基组分别采用GGA-PBE和DNP,内核电子采用DSPP方法处理.范德华力的影响采用了基于Grimme的DFT-D方法进行校正.能量、最大位移和原子间作用力的收敛标准分别为1×10-5Ha、5×10-4nm和0.02 Ha·nm-1.为了加速收敛,采用0.005 Ha的拖尾值.采用Mulliken电荷分布分析电荷转移情况.
CNT和Fe-CNT对MNZ分子的吸附能分别用公式(1)和(2)计算:
Eads=EMNZ-CNT-(EMNZ+ECNT)
(1)
Eads=EMNZ-Fe-CNT-(EMNZ+EFe-CNT)
(2)
在公式(1)和(2)中,EMNZ-CNT、EMNZ-Fe-CNT为复合物的总能量,EMNZ、ECNT分别为药物分子MNZ和CNT的总能量.
能隙(Eg)定义为:
Eg=ELUMO-EHOMO
(3)
其中ELUMO和EHOMO分别为最低未占分子轨道的能量和最高占据分子轨道的能量.HOMO和LUMO能级分别与分子的供电子能力和接受电子能力有关.
3 结果与讨论
3.1 原始CNT对MNZ的吸附
原始(5,5)CNT优化后结构及分子静电势(MEP)如图1所示.MEP图可以对系统中的电荷分布及静电效应进行定性描述.在MEP图中,红色代表了正静电势的区域,蓝色代表了负静电势的区域,白色代表了过渡区.颜色越红表示该区域表面电势越正,反之,颜色越蓝代表该区域表面电势越负.HOMO和LUMO的能量水平为-4.536 eV和-3.664 eV,产生了0.872 eV的能隙,Eg在吸附过程中值的变化可以作为分子稳定性的指标,HOMO-LUMO能隙值越大,说明电子跃迁越不容易发生,分子越稳定.这个相对较小的带隙表明原始CNT可能不能很好的吸附MNZ.
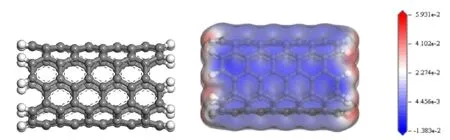
图1 (a)CNT的优化结构和(b)MEP图
MNZ优化后的结构以及MEP图和HOMO图如图2表示,5元环的硝基和羟基及N5原子具有负的静电势,可参与亲核反应.因此,MNZ分子从其带负电荷的中心水平朝向原始CNT.为了研究MNZ在单壁CNT上的吸附行为,共考虑了七种不同的吸附构型并进行优化,其优化结果如图3所示.其中,图3(a)以5元环上连接的甲基上的三个氢原子为一个平面吸附到单壁CNT表面;图3(b)以5元环上连接的硝基上的两个氧原子为一条线吸附到单壁CNT表面;图3(c)以羟基为一个平面并用其中的氢原子吸附到单壁CNT表面;图3(d)以5元环上的碳和其连接的氢原子为一条线并用氢原子吸附到单壁CNT表面;图3(e)以5元环为一个平面吸附到单壁CNT表面;图3(f)以5元环上的氢原子与甲基上的H1原子为一条线吸附到单壁CNT表面;图3(g)以甲基上的C原子和H1原子为一条线吸附于单壁CNT表面.本文对七个不同的吸附模型之间的吸附能、电子轨道、能隙、电荷转移量进行了分析,相关结果如表1所示.从吸附能可以看出,这七种复合结构的吸附能普遍较小.其中,MNZ在甲基、硝基以及五元环上的吸附能力相对较强,而且更倾向于通过甲基被吸附到CNT上(如图3(g)MNZ-CNT-7).从转移电荷值和能隙数据可以看出,在吸附过程中,MNZ得到电荷,而CNT失去电荷,且复合结构的电荷转移量和能隙都普遍较小.此外,对单独的MNZ和CNT以及CNT-MNZ复合物的结构分析表明,MNZ和CNT的几何参数没有明显变化.这些结果表明,原始CNT与MNZ之间的相互作用较弱,且对MNZ分子的吸附效果较差,说明MNZ在原始CNT上的吸附为稳定性较差的物理吸附.因此,原始CNT并不是吸附MNZ分子的理想材料.

表1 原始CNT吸附MNZ分子的吸附能、分子轨道、电荷转移量
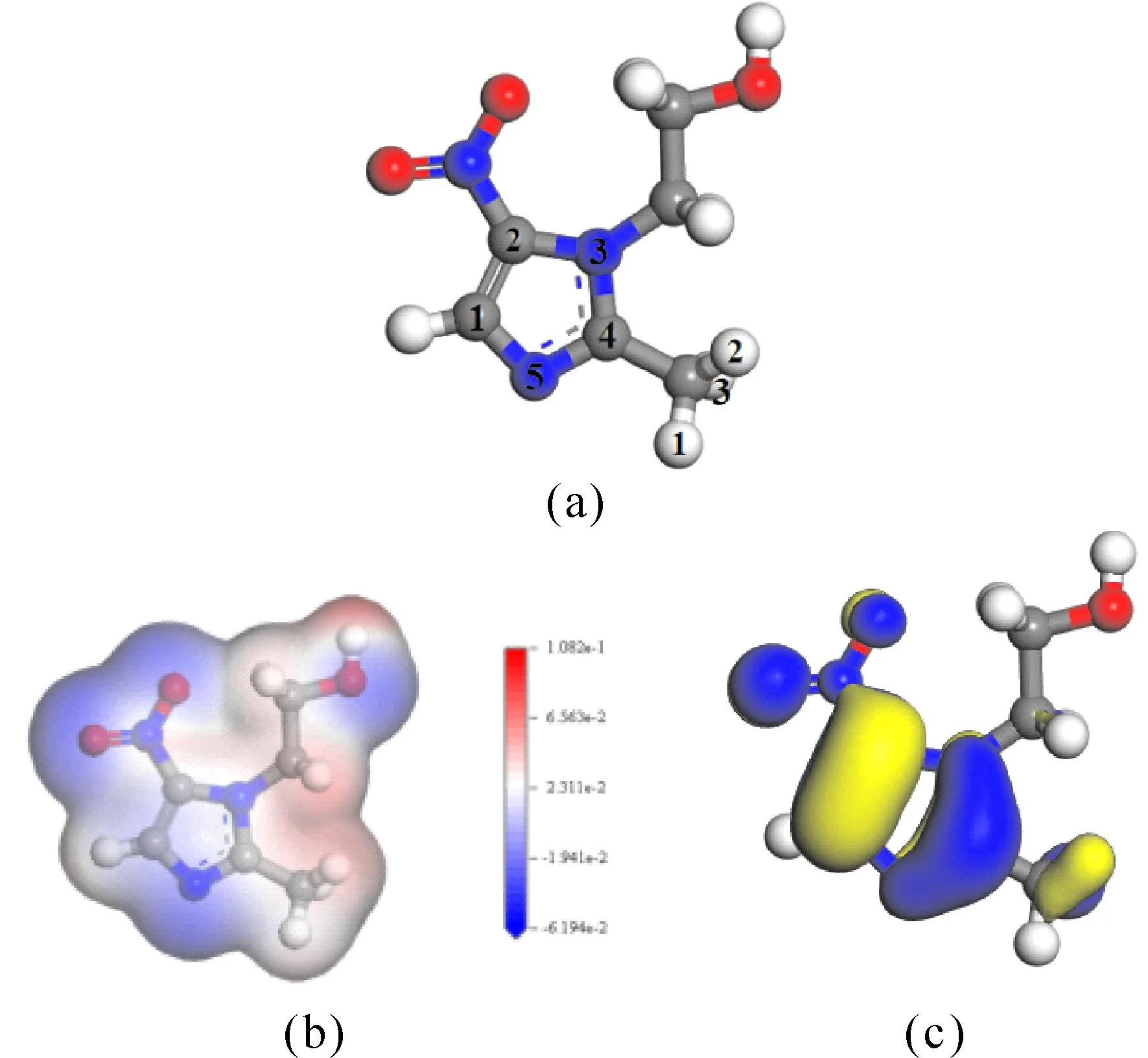
图2 (a)MNZ分子优化后的结构、(b)MEP图和(c)HOMO图
3.2 Fe-CNT对MNZ的吸附
掺杂是改变材料物理化学性质的有效方法,为了得到更为合适的CNT吸附材料,本文采用Fe对CNT进行了掺杂处理:用1个Fe原子替代1个C原子,得到Fe-CNT.图4为Fe-CNT的优化结构及MEP图.其掺杂后的Fe-CNT的带隙为0.505 eV.Fe-C键长的平均值约为1.819 Å,而Fe-CNT的C-C键长约为1.42 Å.与C相比,Fe原子的共价原子半径更大,这导致掺杂点周围的结构发生了变形,使掺杂原子略微凸出平面以减少其较大的共价半径引起的应变,因此MNZ分子吸附的优先位置很有可能是掺杂CNT的结构变形区域.此外,MEP图表明,掺杂的Fe原子具有更多的正静电势,更有可能参与亲电相互作用.
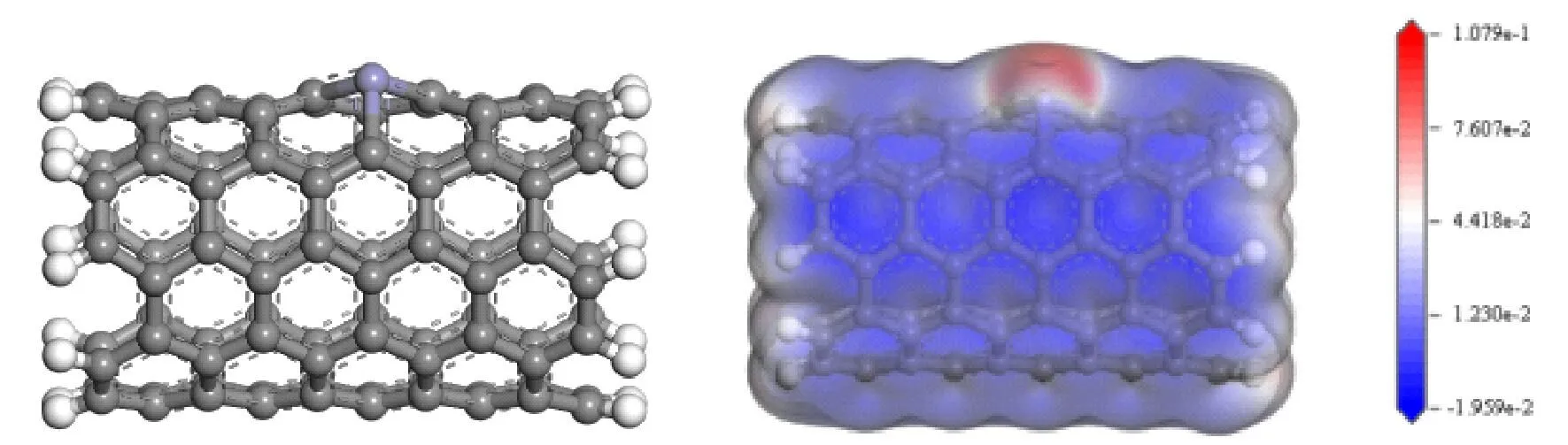
图4 (a)Fe-CNT的优化结构和(b)MEP图
为了研究MNZ分子在Fe-CNT表面上的吸附行为,考虑了与原始CNT表面相同的七种吸附方式去接近Fe-CNT表面的原子,其优化后的结构如图5所示.并分析了吸附能、电子轨道、能隙及电荷转移,相关结果如表2所示.从表2可以发现,Fe-CNT与MNZ的七种复合构型吸附能普遍高于原始CNT.其中,MNZ-Fe-CNT-2、MNZ-Fe-CNT-4和MNZ-Fe-CNT-5的吸附能相对较高,且MNZ-Fe-CNT-2的吸附能最高,说明MNZ更倾向通过其-NO2基团被吸附到Fe-CNT上.在MNZ-Fe-CNT最稳定复合物中(如图5(b)MNZ-Fe-CNT-2),Fe-O相互作用距离的平均值约为2.247 Å.从表2还可以看出,吸附能为-2.277 eV,与MNZ-CNT-2的吸附能-0.787 eV相比,Fe-CNT对MNZ分子的吸附效果大大增强;能隙为0.393 eV,与MNZ-CNT相比能隙显著减小且能隙变化量从1.49%增加到22.18%,说明Fe-CNT对MNZ的吸附作用明显增强且稳定性更高;电荷转移量为0.087e,这说明电子从MNZ分子转移到了Fe-CNT表面,电荷转移量明显升高.同时N-O键长分别从1.241 Å和1.242 Å增加到了1.282 Å和1.310 Å,O-N-0的键角由124.327°变为114.318°.键长和键角都发生了明显的变化,说明Fe原子掺杂后对MNZ的结构产生了一定的影响,MNZ与Fe-CNT之间的相互作用显著增强.MNZ分子在Fe-CNT上的吸附由物理吸附变为化学吸附.因此,Fe-CNT是吸附MNZ分子的理想材料.

表2 Fe-CNT吸附MNZ的吸附能、分子轨道和电荷转移量
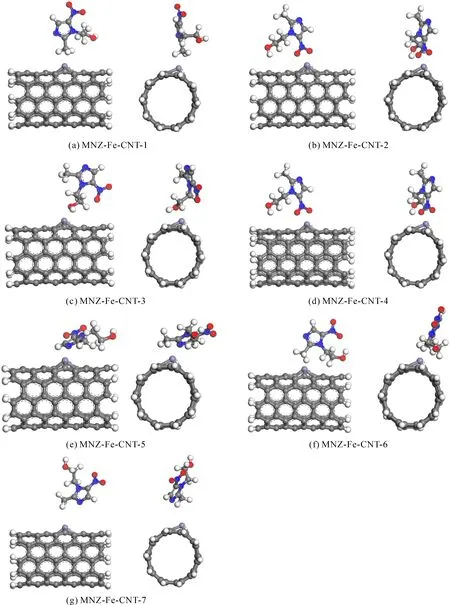
图5 Fe-CNT吸附MNZ的七种构型
为进一步研究Fe-CNT对吸附MNZ分子的吸附机理,计算并分析了MNZ-Fe-CNT-2构型的态密度,结果如图6所示.从图6(a)的态密度图可以看出,MNZ-Fe-CNT体系与MNZ-CNT体系相比,在-15~-10 eV附近峰值明显增加.此外,在掺杂Fe原子后,其态密度在费米能级附近的值升高,带隙也变小,这造成了电子在价带和导带之间更容易发生转移,表明CNT在掺杂了Fe原子后与MNZ分子的相互作用增强.在图6(b)中,Fe原子的3d轨道和O原子的2p轨道在-27.5~-25 eV、-22.5~-20 eV、-12.5 eV和-5~-2.5 eV的附近存在明显的重叠,轨道之间出现杂化现象,杂化轨道比原来轨道的成键能力强,说明掺杂Fe原子后的CNT与MNZ分子形成的结构体系更加稳定.
4 结 论
本文采用了第一性原理计算方法分别研究了原始CNT和Fe-CNT对MNZ分子的吸附行为.主要研究结果如下:
(1)原始的CNT对MNZ的吸附作用较差,属于物理吸附,不适合做MNZ分子的吸附材料.
(2)与原始CNT相比,Fe-CNT对MNZ分子的吸附作用明显增强,且电荷转移量明显增加.Fe-CNT有望成为吸附水中污染物MNZ的候选材料.
