家族性ATM基因纯合突变共济失调毛细血管扩张症家系及文献复习
2023-12-21袁洁王艳艳
袁洁,王艳艳
患儿,男,7岁。主因“走路不稳4年余,咳嗽10余天,咯血2天”于2013年6月10日入院。4年前患儿无明显诱因出现走路不稳,进行性加重,未给予特殊治疗。入院前1年曾患肺炎,对症治疗痊愈。入院10余天前患儿出现阵发性咳嗽,无痉挛性咳嗽,无昼夜规律,痰不多,无喘息和呼吸困难,就诊当地医院,给予对症治疗(具体不详),患儿咳嗽无明显减轻;入院前2天突然出现咯血,呈暗红色,量约20 ml,病程中体温最高37.8℃,无盗汗、乏力及明显消瘦,当地医院查血常规示WBC 20.34×109/L, NE 83.8%, Hb 114 g/L, PLT 557×109/L。胸部X线片示肺炎,腹部B型超声显示脾稍大,凝血功能基本正常,家长为进一步治疗就诊入院。自发病以来,患儿精神反应可,饮食睡眠正常,尿便无异常。既往无反复呼吸道感染史。患儿系G4P4,足月顺产,出生史无异常。婴幼儿期生长发育史正常,3岁后逐渐出现共济失调,其父母均健康,非近亲结婚。第一胎为男孩,11岁时因“肺炎”夭折,第二、三、五胎均为女孩,均存在走路不稳的情况。入院查体:T 37℃,P 96次/min,R 28次/min,BP 120/70 mmHg,末梢血氧饱和度(SaO2,未吸氧)99%。神志清楚,反应可,全身皮肤无皮疹及出血点,可见卡介苗瘢痕,浅表淋巴结未触及肿大,双眼球结膜可见毛细血管扩张(图1),口唇无发绀。呼吸平稳,两肺呼吸音粗,未闻及明显干湿性啰音。心音有力,律齐,各瓣膜听诊区未闻及病理性杂音。腹软,无压痛及反跳痛,肝脾肋下未触及。四肢末梢暖,肌力正常,肌张力增高,腱反射存在,步态不稳,指鼻、指指及跟膝胫试验(+)。血常规:WBC 7.4×109/L,NE 58.5%,Hb 103 g/L,PLT 458×109/L。CRP 24.1 mg/L,肝肾功能及心肌酶正常。肺炎支原体抗体阴性。甲胎蛋白阳性。便常规正常,便潜血阳性。脑脊液:常规及生化无明显异常。免疫球蛋白测定:IgG 0.03 g/L(正常参考值5.72~14.74 g/L),IgA 0.05 g/L(正常参考值0.34~3.05 g/L),IgM 1.25 g/L(正常参考值0.31~2.08 g/L)。T淋巴细胞免疫功能:CD4+24.7% (正常参考值35%~58%),总B细胞1.9%(正常参考值5%~18%),CD3+、CD8+、CD4/CD8正常,自然杀伤淋巴细胞23.4%(正常参考值6%~20%)。胸部CT:双侧支气管肺炎伴双下肺支气管扩张,胸腺未见异常(图2)。头颅MR:考虑双侧小脑半球萎缩,双侧乳突、双侧上颌窦、蝶窦、左侧筛窦炎性改变(图3)。入院后给予抗感染、止血及免疫球蛋白等对症支持治疗,患儿咯血减轻,病情好转出院。出院后继续随访,患儿多次因呼吸道感染于当地住院治疗,2019年10月因呼吸道感染死亡。
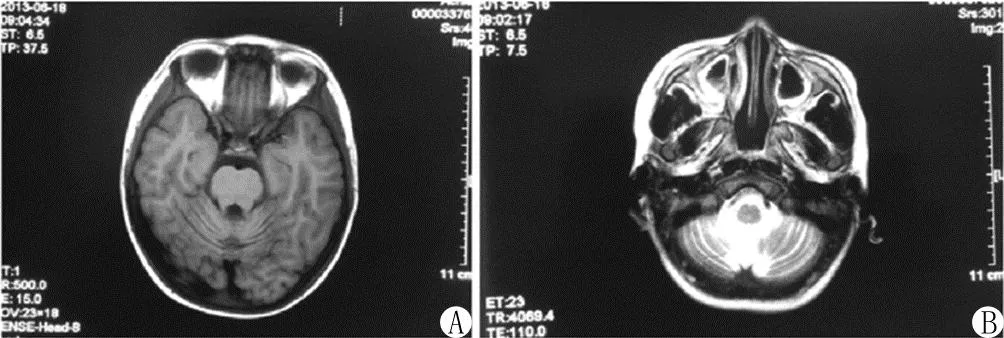
注:A.小脑萎缩;B.鼻窦炎
基因检测结果(图4):患儿ATM c.6100C>T,p.(Arg2034*)纯合突变,患儿父亲及母亲均为ATM c.6100C>T,p.(Arg2034*)杂合突变,患儿姐妹均为ATM c.6100C>T, p.(Arg2034*)纯合突变。由于经济原因,患儿祖父母及外祖父母未查相关基因检测。遂诊断该患儿为共济失调毛细血管扩张症。
家系随访(图5):家系中共有5个子女,2男3女,先证者为第4胎。所有儿童出生史均无异常,婴幼儿期均正常学步走路。3~4岁时逐渐出现共济失调,至学龄前期开始出现反复肺部感染,均有咯血表现,由痰中带血到大块鲜血。长子11岁时夭折,女孩寿命均在11~13岁。先证者13岁死亡,全部子女均死于反复肺部感染,未发现肿瘤。患儿父母目前均身体健康,未发现毛细血管扩张、共济失调表现及肿瘤等,祖父母及外祖父母因高龄步态不稳,但无毛细血管扩张表现及肿瘤发现。
讨 论共济失调毛细血管扩张症(ataxia telangiectasia, A-T)是通过常染色体隐性方式遗传的一种少见多系统疾病,以进行性小脑共济失调为特征,常累及神经系统、血管、皮肤、网状内皮系统及内分泌系统等[1-2]。目前发病率为1/100 000~1/40 000[1,3]。其发病机制与ATM基因突变有关。ATM定位于人类染色体上11q22-23,由66个外显子(4个非编码区和62个编码区)组成,长度150 kb,编码一个转录区,其编码产物为ATM蛋白,存在于细胞核和细胞质中,可协调细胞信号通路,维持细胞氧化还原平衡[4]。
A-T患者的临床表现复杂多样,临床表型与基因表型密切相关。临床特征包括:婴幼儿期发病的小脑共济失调、眼球结膜毛细血管扩张、反复发作的鼻窦炎和肺部感染,易发生恶性肿瘤和混合性免疫缺陷病[4]。
现用“经典”和“变异”来区分A-T的2种不同但被广泛认可的共济失调表现。在经典表型中,共济失调首先出现在幼儿阶段,通常在学步期正常行走,随后出现行走倒退并进行性加重,早期易被误诊为共济失调性脑瘫。在小学阶段,逐渐进展至行走困难,常需要轮椅辅助行走。大多数神经系统问题在12~15岁不再进展。大部分在20岁前需要轮椅辅助者,很难活过30岁。在变异表型中,小脑共济失调通常是轻症或不存在的,相反,锥体外系运动障碍,如肌张力障碍和震颤是主要表现[1]。
球结膜内毛细血管扩张通常发生在5~8岁,有时更晚或根本不出现。毛细血管扩张也可出现在暴露的皮肤区域,尤其是面部和耳朵。球结膜毛细血管扩张不会影响视力[5],但由于眼球运动的协调性受损,会影响需要快速、准确眼球运动的功能(如阅读)及精细运动功能(如书写、绘画和进餐),并可能出现构音障碍[1]。本例先证者及其兄长、姐妹均在学步期生长发育正常,3岁后逐渐出现共济失调且进行性加重,有球结膜毛细血管扩张,存在ATM基因异常,是经典表型。由于家系中5个子女均学龄期死亡,长期住院治疗,无法评价其学习能力。
大约2/3的A-T患者有免疫异常,最常见的是1种或多种低免疫球蛋白(IgG、IgA、IgM或IgG亚类)和淋巴细胞减少症,特别是影响T淋巴细胞[1,6]。可导致反复肺炎、支气管扩张和间质性肺病(ILD)。本例先证者出现IgA和IgG极低及T淋巴细胞减少,并有反复呼吸道感染表现,符合经典表型。
A-T患者的癌症发病率明显增加,淋巴瘤和白血病最常发生在20岁以下经典型A-T患者,而成年人多为淋巴瘤和实体瘤(包括乳腺癌、肝癌、胃癌和食管癌)[1]。目前尚无可靠的方法预测淋巴瘤和白血病的发生。儿童患者虽然血液系统肿瘤多发,但尚未检索到引起血小板数量异常的相关报道。仅1981年报道1例A-T患者表现为严重的反复出血,其特征是出血时间延长,血小板聚集缺失,但血小板计数和血块回缩试验正常[7]。先证者就诊时血小板升高(458×109/L),出院前复查下降(309×109/L),考虑与患儿当时的呼吸道感染有关。研究证实,急慢性炎性疾病过程中可发生血小板数量改变及活化,血小板—中性粒细胞复合物数量增加可预示疾病的严重程度[8]。本家系中5个患儿寿命均未超过14岁,无肿瘤发生。随着年龄的增长,进食和吞咽困难逐渐导致营养不良,是A-T的常见特征。营养不良、感染、生长因子及激素水平缺乏被认为是导致生长发育迟缓的原因,女性比男性更突出[1,9]。少数A-T患者在疾病晚期出现胰岛素抵抗型糖尿病[10]。A-T患者也可出现早衰特征,如少年白头,也可有广泛且难以治疗的白癜风等[1]。
A-T的神经病理学标志是小脑半球的弥漫性变性或萎缩,涉及浦肯野细胞(PC)颗粒神经元,磁共振成像(MR)是观察中枢神经系统(CNS)的首选方式[1,11]。本例患儿头颅MR出现脑萎缩及鼻窦炎改变与A-T的影像特点相符。据报道,ATM杂合突变者对辐射敏感性较正常人敏感;但与纯合突变者比较,后者更敏感。因此宜减少放疗及射线暴露次数[9,12]。
超过25%的A-T患者存在慢性肺部疾病[1,13],一旦发生感染,可能增加呼吸道症状的严重程度,导致支气管扩张、肺部纤维化和ILD等严重的肺部疾病。临床上应早期关注A-T患者的呼吸道症状,一旦出现反复咳嗽、胸闷和/或喘息,应高度重视,积极治疗。A-T患者由于肌肉协调受损导致呼吸肌无力,出现肺功能下降,表现为限制性通气功能障碍,其特征是用力肺活量(FVC)减低,增加手术麻醉意外风险,如需手术应做好术前评估,术中密切监测。纤维支气管镜技术作为临床药物治疗的辅助手段,早期为A-T患者行支气管镜肺泡灌洗术,可以减轻肺部炎性反应,查找病原,提供精准治疗。
A-T是一种复杂的疾病,患者虽然有相同的症状但在疾病进展和并发症发生率方面各有不同。临床诊断主要依据神经系统改变即小脑共济失调,结合以下1种或多种症状:毛细血管扩张、免疫缺陷致反复呼吸道感染及辅助检查结果异常,包括IgA缺乏、T淋巴细胞显著减少、血清AFP升高及头颅MR显示小脑萎缩。基因诊断是确诊的金标准。有报道17个月大即出现发育迟缓表现,3岁开始出现共济失调[14],而本例先证者3岁前生长发育均正常,3岁后出现共济失调、有球结膜毛细血管扩张、免疫缺陷及甲胎蛋白异常,存在ATM基因改变,诊断明确。追踪本家系5个患儿均在婴幼儿期正常生长发育,3~4岁开始出现行走倒退并进展迅速,神经系统症状出现后反复下呼吸道感染相继发生,全部于11~14岁间因感染去世,无肿瘤病例发生。患儿父母虽非近亲结婚,但同为杂合突变,后代5个子女均为纯合突变的经典A-T患者,概率很低但真实发生,经过随访,家系中的所有儿童均已过世,说明纯合突变病情更重,预后更差。
A-T的管理和治疗以对症和支持治疗为主,目前尚无根治方法。在经典型A-T表现中,共济失调首先出现,最早可发生在幼儿阶段,进行性加重,存活时间与神经系统损害严重程度并不显著相关[1],目前尚无特效减缓或停止神经退行性变的治疗方法[11]。一项没有安慰剂对照的试验表明,金刚烷胺、多巴胺激动剂和N-甲基-D-天冬氨酸(NMDA)拮抗剂,可能会缓解共济失调,有文献证实类固醇激素可以暂时改善共济失调[15-16],但考虑到不良反应应谨慎应用。经颅多普勒超声小脑刺激可能是一种很有前途的治疗方法[17]。
预防感染和提高免疫力是治疗关键。有报道大剂量免疫球蛋白静脉滴注、胸腺肽肌内注射可提高患儿的免疫功能[18],但有待于进一步统一认识。在变异型A-T中,肌张力障碍和震颤是主要表现,可以通过康复技术维持或改善活动质量。考虑到该疾病的复杂性和严重性,一旦怀疑应尽早确诊,重点关注呼吸系统症状,早期干预治疗,避免错过最佳治疗时机。
(致谢:感谢河北省儿童医院安淑华教授对本文撰写提供的无私帮助及指导,感谢河北省儿童医院李金英主任医师、邯郸市第二医院张亚黎主任医师对本文资料收集提供的大力支持)
