新复合杂合基因突变致非经典21-羟化酶缺陷症1例报告并文献复习
2023-12-06祁梦梦王雪梅刘云婷王倩辛倩玉林华吕文山杨丽丽
祁梦梦 王雪梅 刘云婷 王倩 辛倩玉 林华 吕文山 杨丽丽
(1 青岛大学附属医院内分泌与代谢性疾病科,山东 青岛 266003; 2 青岛大学附属医院内科学教研室;3 北京迈基诺基因科技股份有限公司医学检验所; 4 青岛大学附属医院市南院区门诊)
21-羟化酶缺陷症(21-hydroxylase deficiency,21-OHD)是先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)的最为常见类型,大约占95%[1-2]。21-OHD因皮质醇(CORT)合成所必需的酶缺陷导致17-羟孕酮、雄烯二酮、孕酮等性激素水平升高所致[3]。21-OHD可进一步分为经典型和非经典型,其中非经典型临床表现较轻,容易被误诊和漏诊。但非经典型的发病率在21-OHD临床谱中最高,普通人群的发病率高达0.1%[4]。在雄激素过多的女性中,非经典型21-OHD的全球患病率更高达4.2%[5]。非经典21-OHD患者多具有多毛症、闭经、多囊卵巢综合征、月经紊乱和不育等的临床特征[6-7],如父母双方均为非经典21-OHD患者,后代患严重21-OHD的概率更高[8-9]。本文对1例非经典型21-OHD中的迟发型患者的表型和基因型进行分析,以期提高临床医师对该病认识。
1 临床资料
患者,女,16岁。家长发现患者10岁时面部出现小须,十二三岁时身高增长快速,但自此以后身高增长速度减缓。患者13岁时因面部出现痤疮、乳腺未发育、无月经来潮于他院就诊,检查示促肾上腺皮质激素(ACTH)19.97 ng/L,CORT 416.8 nmol/L,染色体核型分析为46,XX,考虑为21-OHD可能,建议完善相关基因检测以明确诊断,患者及家属未积极配合。
2021年2月,患者因多毛6年、面部痤疮3年、乳腺未发育以及无月经来潮来我院就诊,门诊以CAH收入内分泌科。体格检查:神志清,精神可,面色黝黑,面部有胡须、痤疮。甲状腺Ⅱ度肿大。乳房Ⅰ期。阴毛Ⅲ期,阴蒂肥厚。
入院后完成电解质及性腺、甲状腺、肾上腺以及垂体相关激素等检查。其中,性激素测定显示泌乳素644.70 mU/L,雌二醇722.60 pmol/L,促黄体生成激素1.07 U/L,促卵泡生成素3.14 U/L,孕酮13.58 nmol/L,睾酮16.35 nmol/L,血17α-羟基孕酮 91.51 μg/L,血脱氢表雄酮 11.85 μg/L。甲状腺功能检查示抗甲状腺球蛋白抗体<10.00 kU/L,促甲状腺激素2.68 mU/L,游离三碘甲状腺原氨酸为6.97 pmol/L,游离甲状腺素19.50 pmol/L,抗甲状腺过氧化物酶抗体<9.00 kU/L。肾上腺和垂体相关激素:ACTH(8:00)54.49 ng/L,CORT(24:00)45.4 nmol/L。鞍区(垂体)MR平扫+单脏器薄层扫描示垂体饱满。肾上腺CT薄层平扫(轴位)示双侧肾上腺增粗,考虑肾上腺皮质增生可能性大;经腹部多普勒超声检查示子宫轮廓偏小;右侧卵巢约4.0 cm×1.6 cm大小,内部回声未见异常;左侧卵巢约2.4 cm×0.9 cm大小,内见一约2.1 cm×1.1 cm大小较大卵泡。根据患者的临床症状、体格检查以及辅助检查等结果,考虑为CAH。
为明确诊断,指导治疗,采集患者血液,行全外显子测序,结果显示CYP21A2基因有复合杂合突变[错义突变c.1451G>A(p:R484Q),缺失突变deletion exon 1~3],这种复合杂合突变以前未见相关文献报道,患者确诊为非典型21-OHD。征得其亲属同意,采集外周血针对CYP21A2进行高通量基因测序,结果显示,其外祖父及母亲携带c.1451G>A(p.R484Q)等位基因,而其祖母、父亲及弟弟均为deletion exon 1~3号外显子杂合缺失携带者(图1)。
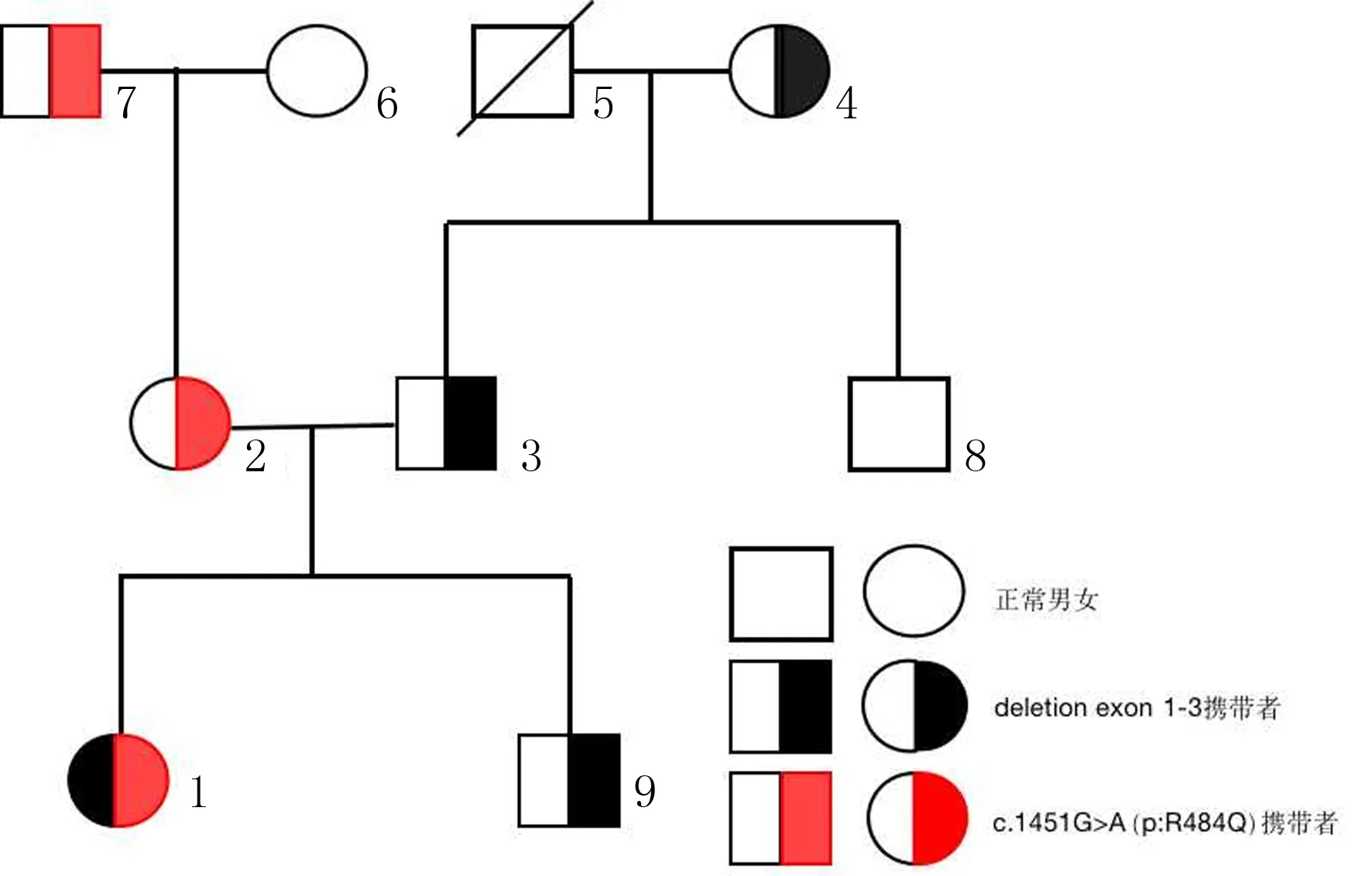
1:先证者,2:先证者母亲,3:先证者父亲,4:先证者祖母,5:先证者祖父,6:先证者外祖母,7:先证者外祖父,8:先证者父亲哥哥,9:先证者弟弟
患者给予地塞米松0.75 mg 每日1次替代治疗,次日复查ACTH以及CORT,结果显示ACTH2.21 ng/L,CORT 18.60 nmol/L。患者状态良好,调整地塞米松用量为0.375 mg,每日1次,于次日再次进行ACTH及CORT复查,结果显示ACTH 73.30 ng/L,CORT 76.20 nmol/L,调整地塞米松用量为0.75 mg 每日1次。患者病情改善,出院治疗。患者出院后定期复查性激素6项及ACTH、CORT水平,及时调整治疗方案。患者于规律服药2个多月后月经来潮,后患者月经规律,周期约为28 d,并且患者面部多毛、痤疮问题明显改善。
2 讨 论
CAH是一组异质性先天性疾病,该病的发生主要是由肾上腺皮质激素合成中酶缺陷所致CORT合成不足,继发下丘脑分泌促肾上腺皮质释放激素和垂体ACTH代偿分泌增加所造成。CAH属常染色体隐性遗传病,其中21-羟化酶缺乏最常见,约占95%以上。21-OHD是导致新生儿生殖器官缺陷的最常见原因,可根据患者17-羟孕酮的水平分为经典型和非典型[10-12]。经典型又可进一步分为失盐型和单纯男性化型[13-14]。经典型患者具有肾上腺功能不全并伴有不同程度的男性化的典型临床表现,血清17-羟孕酮水平超过242 nmol/L;而非典型患者的临床症状不典型,血清17-羟孕酮水平也在正常水平之内,常被误诊为多囊卵巢综合征[2]。非经典21-OHD以青春期前出现多毛、痤疮、身体生长加速和骨龄提前为典型表现,其中女性患者很容易被漏诊和误诊[6,15]。本例患者即为长期漏诊的非经典型21-OHD的迟发型。随着分子诊断技术的进展,目前研究发现,在高雄激素的患者中非经典21-OHD的患病率高达2.5%[16]。因此,应提高对女性青春后期高雄激素者的关注,及时诊断和治疗,以避免漏诊和误诊。比如本例患者就诊时已经处于青春后期且骨骺已经闭合,提示及时的诊断和治疗可以提高21-OHD患者的身高[16-17]。
21-OHD患者临床表现差异很大,常需基因检测才能确诊[18]。本例患者的基因检测结果显示,其CYP21A2基因存在exon 1~3杂合缺失和杂合错义突变[c.1451G>A(p.R484Q)]。经由家系验证,exon 1~3缺失遗传自父亲。该突变最早由NOR-DENSTRÖM等[19]报道,本例患者为CYP21A2基因1~3外显子纯合缺失,该缺失可致CYP21A2蛋白富含疏水氨基酸亮氨酸的N端及部分螺旋结构缺失,致使21-羟化酶活性完全缺失,可导致患儿死亡[20]。经询问家族史,其母亲曾生育2名婴儿,但因顽固性呕吐和严重电解质紊乱伴高17-羟孕酮血症,分别于生后第40、50 天死亡。CYP21A2基因c.1451G>A(p.R484Q)最早由SPEISER等[21]报道,他们先后发现一对疑似21-羟化酶中度缺乏致阴茎/阴蒂早期发育以及阴毛增长迅速的兄妹。基因检测结果显示患儿存在c.518T>A(p.I173N)和c.1451G>A(p.R484Q)复合杂合突变。ONO等[22]发现,R484与CYP21A2蛋白J螺旋的D322位点相互影响,且在A-螺旋中具有稳定偶极螺旋结构的作用。通过结合液相色谱-电喷雾串联质谱技术对体外细胞进行相关检测,结果发现c.1451G>A(p.R484Q)突变细胞株CYP21A2酶活性降至野生型的3.8%[23]。因此,SPEISER等[21]报道的先证者兄妹携带的两种错义突变所产生的蛋白均保留一定的酶的活性,与生化实验室所测其21-羟化酶中度缺乏结果相符。21-OHD是一种常染色体隐性遗传病,其表型取决于CYP21A2酶残存活性。
与其他先天性疾病不同的是,CAH目前已经有确切的药物进行治疗,甚至可以进行产前治疗[24]。2018年The Endocrine Society(TES)针对该病发布了第二版《类固醇21-羟化酶缺乏导致的先天性肾上腺皮质增生症指南》。该指南建议不能仅依靠彩超进行性别诊断,为保险起见,还需对母亲血液进行Y染色体DNA的基因筛查,确诊为21-羟化酶缺乏的女性胎儿,再对其进行治疗[25]。对于经典型患者,从婴儿时期即给予盐皮质激素和氯化钠治疗,有助于减少糖皮质激素的治疗剂量,并维持患儿正常生长。无症状的非经典患者可以不予治疗,但若有骨龄提前、性早熟、明显男性化、不孕或不育等表现,也建议予以糖皮质激素治疗。对于非经典21-OHD女性患者来说,如果替代治疗时用药不规律,或盲目停止用药,会使患儿男性化程度进一步加重[26]。
综上所述,本文通过对1例非典型21-OHD中的迟发型患者的诊疗经过进行回顾,并进行相关文献复习,发现了CYP21A2基因一种新的复合杂合突变,进一步丰富了21-OHD致病突变数据库,有助于提高临床医师对该病的认识,避免此类疾病的漏诊和(或)误诊。早期确诊并对患者进行规范治疗,可以显著改善患者预后。
作者声明:祁梦梦、王雪梅、吕文山、杨丽丽参与了研究设计;祁梦梦、刘云婷、王倩、辛倩玉,林华参与了论文的写作和修改。所有作者均阅读并同意发表该论文,且均声明不存在利益冲突。
