CRPPA基因变异导致肢带型肌营养不良症1例☆
2023-10-30杨媛媛刘蕊范宽韩璐刘嘉鹏李世容瞿浩王添阳刘坤雪灵胡晓
杨媛媛 刘蕊 范宽 韩璐 刘嘉鹏 李世容 瞿浩 王添阳 刘坤雪灵 胡晓
肢带型肌营养不良症(limb-girdle muscular dystrophy,LGMD)是一组具有异质性的遗传性肌肉疾病,主要表现为进行性四肢近端无力、肌酶升高、腓肠肌肥大和Gowers征[1-3]。LGMD可分为常染色体显性遗传型(LGMD1型)、常染色体隐性遗传型(LGMD2型)和X染色体遗传型[4]。至今已有39个基因被发现与LGMD相关,但许多相关变异的致病性尚不明确[5-6]。不同基因变异导致的不同亚型在临床表现、病程和预后上也具有明显的异质性,然而目前对其中的表型-基因型相关性的了解仍然有限[7-8]。本文报告1例由CRPPA基因变异引起LGMD患者,分析了其基因型和表型,并进行文献复习。
1 临床资料
患者,男,21岁,主因“双下肢进行无力7年,加重2年”入院。患者近7年逐渐出现双下肢无力,行走时明显,须蹲下休息片刻方能继续行走,下蹲后自行站起困难,近2年双下肢无力程度逐渐加重。既往史无特殊。家族史:患者父母否认近亲结婚及特殊家族史(图1)。
入院查体:神志清楚,言语流利,无精神智能障碍。脑神经检查无明显异常。四肢肌张力对称适中,颈屈肌、双上肢肌力5级,髂腰肌肌力2级,双下肢股四头肌肌力5-级、股二头肌肌力4+级、足背屈肌力4级,跖屈肌力4级。无共济失调。四肢腱反射对称(+),全身深浅感觉正常。双侧病理征阴性。颈软,克氏征、布氏征阴性。
肌酶检查:乳酸脱氢酶284 U/L(参考值:120~250 U/L),肌酸激酶4410 U/L(参考值:50~310 U/L),肌酸激酶同工酶122 U/L(参考值:0~15 U/L),α-羟丁酸脱氢酶176 U/L(参考值:80~220 U/L)。肌电图:所检肌肉均可见自发点活动,四肢近端运动单位电位波幅下降,相位增多、时限缩窄;所检测神经的运动神经传导速度、感觉神经传导速度及F波正常范围;符合典型肌源性损害表现(图2)。双下肢肌肉磁共振:双侧大腿、小腿多发肌群信号异常,信号不均匀,内见“雪花”样长T1稍长T2信号,压脂序列呈高信号,肌间隙尚清晰(图3)。骨骼肌活检:组织电镜下可见大部分肌细胞肌原纤维结构无明显病变,少数肌细胞局灶性肌丝溶解;肌细胞胞浆内肌浆网轻度扩张,小脂滴轻度增多,散在或短串状分布;可见少量萎缩肌细胞,未见明显坏死肌细胞,肌细胞间隙轻度增宽,可见局灶性束状胶原纤维沉积;未见明显炎性细胞浸润,小血管扩张、淤血(图4)。由于患者肌活检标本量有限,未能完成HE及其他特殊染色。通过全外显子组基因测序及变异注释和筛选,考虑患者所携带的CRPPA基因纯合变异NM_001101426.3:c.1114_1116delGTT为该家系致病变异(图5)。该变异导致氨基酸发生整码变异p.V372del。经家系验证分析,患者父母携带该位点杂合变异(图1、5)。
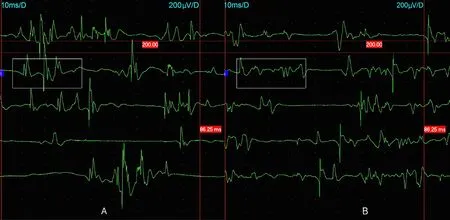
图2 肌电图结果

图3 双下肢MRI检查结果

图4 肌活检电镜结果
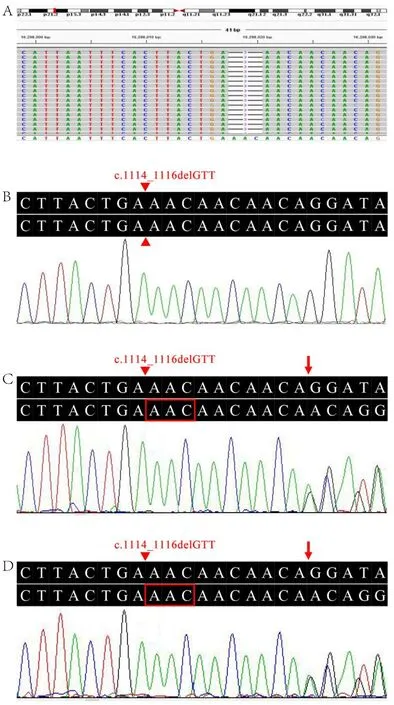
图5 患者及其父母CRPPA基因测序结果
gnomAD数据库中CRPPA基因c.1114_1116delGTT变异频率为0.00007262。SIFT、MutationTaster和CADD均预测该变异为有害或者致病性。既往已有11例该变异所致隐性遗传性LGMD病例报告[9-11]。ClinVar数据库对该变异的致病性分析为致病性或疑似致病性。对野生型和变异型CRPPA蛋白的结构分析表明,该变异导致第372号氨基酸缺失,并导致第367位与364位氨基酸间新增氢键,改变了蛋白质构像(图6)。参照ACMG遗传变异分类标准与指南,该变异可被确定为致病性变异(PM3_强+PM2+PM4+PP1+PP3)[12]。据此,该患者可诊断为C7型MDDG(MDDGC7,OMIM:616052)。住院期间给予患者“辅酶Q10”口服及对症康复治疗效果欠佳。出院半年后随访患者上下肢无力症状加重不明显。
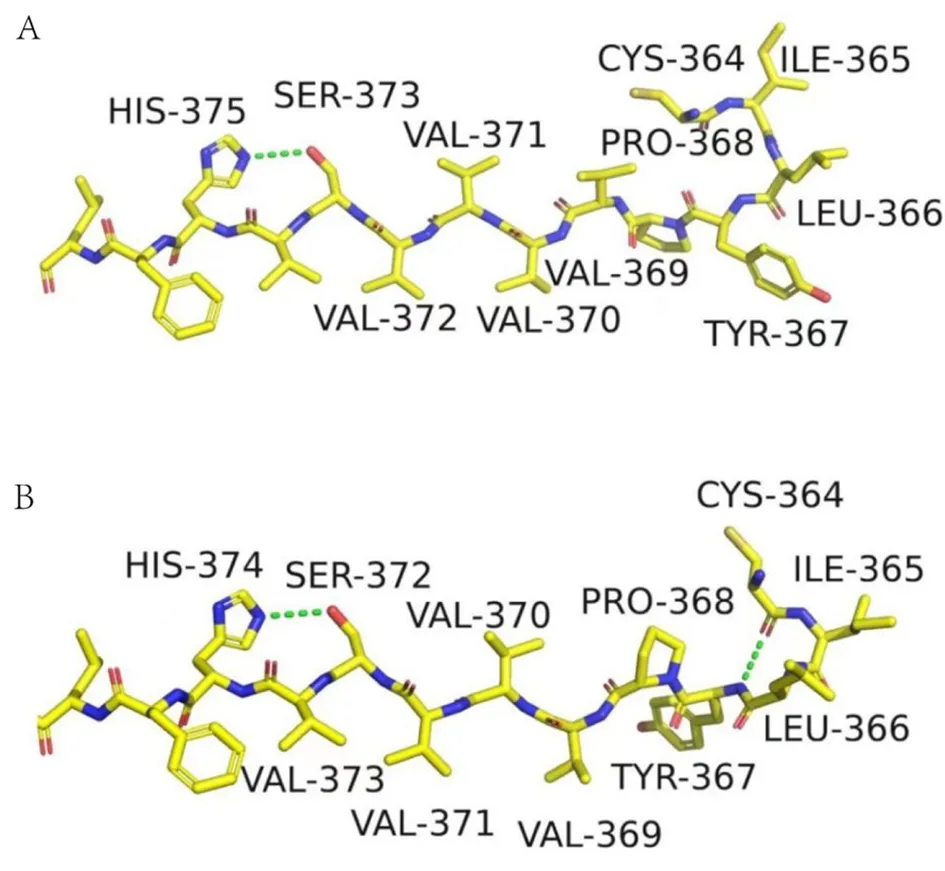
图6 CRPPA蛋白结构分析预测结果
2 讨论
在中国知网中未检索到CRPPA基因相关病例。经检索和筛选,在PubMed数据库中共报告64例MDDG7,其中中国报告8例[9-11,13-19]。64例MDDG7患者的临床表型包括宫内生长受限、腹裂、Walker-Warburg综合征、LGMD和鹅卵石无脑畸形,其中MDDGC7患者24例。MDDG7患者主要临床表现包括手臂和腿部肌肉无力和萎缩,常累及近端肌肉;部分患者会出现关节挛缩或小腿肌肉肥大,血清中肌酸激酶升高;查体可有腓肠肌肥大和Gowers征。其中有部分患者可出现眼和中枢神经系统症状,还可能影响呼吸及心脏功能。部分患者在青少年时期就失去行走能力甚至死亡,也有部分患者临床症状可一直较轻。有报道使用泼尼松治疗MDDGC7患者有一定疗效,但停药后症状加重,暂无其他临床有效的治疗方案[11]。
α-DG是肌营养不良蛋白糖蛋白复合物高度糖基化的核心成分,其依赖充分的糖基化以在肌膜和细胞外基质之间形成连接[20-21]。目前发现参与α-DG糖基化的基因包括:POMT1、POMT2、POMGNT1、POMGNT2、B3GALNT2、POMK、FKTN、FKRP、LARGE、RXYLT1、B4GAT1、GMPPB、DPM1、DPM2、DPM3,DAG1、DOLK和CRPPA[22-23]。这些基因的致病性变异可以导致α-DG糖基化功能障碍并引起多种肌营养不良症[24]。这类疾病具有明显的临床和遗传异质性,但均可在患者肌肉活检中发现标志性的α-DG糖基化减少,故被统称为MDDG。MDDG根据临床表现的不同可分为表现为先天性肌营养不良伴脑部和眼部异常(严重者亦称为Walker-Warburg综合征)的MDDGA、表现为先天性肌营养不良不伴智力发育受损的MDDGB和仅表现为LGMD的MDDGC;根据致病基因的不同也可以分为1-15不同亚型[25-26]。
CRPPA基因编码胞苷二磷酸(cytidine diphosphate,CDP)-L-核糖醇焦磷酸化酶A。该酶合成CDP-核糖醇,为FKTN和FKRP基因编码蛋白糖基化α-DG提供供体底物[20,27]。该基因变异可导致α-DG功能障碍从而引起MDDG7,包括LGMD2U、Walker-Warburg综合征和肌肉-眼-脑疾病[28]。在MDDG7患者的成纤维细胞和骨骼肌活检中,CDP-核糖醇浓度降低,表明CRPPA基因致病变异可导致CDP-核糖醇合成不足,进而引起α-DG糖基化障碍和肌营养不良。有报道提出患者血液中CDP-核糖醇含量检测可能是诊断并评估该病疗效的有效方法,且补充核糖醇可以恢复成纤维细胞中CDP-核糖醇的浓度,膳食核糖醇治疗对MDDG7患者可能有效,然而尚缺乏进一步验证[27]。
综上所述,MDDG和LGMD均具有较强的临床和遗传异质性,且早期临床特征往往不典型,难以与其他肌病鉴别。目前的遗传学技术已能为相当数量的MDDG患者明确诊断。该类疾病应尽早行基因检测明确诊断并进行遗传咨询,以方便患者家庭治疗管理,并通过产前诊断终止遗传链,避免本病患儿出生以减轻家庭及社会的负担。该病目前尚缺乏有效的治疗方案,补充核糖醇可能是其治疗的新研究方向,CDP-核糖醇检测可能是诊断和评估核糖醇饮食治疗效果的有效方法[29-30]。本文报告了1例仅表现为双下肢无力的MDDGC7病例,发现患者携带CRPPA基因c.1114_1116del纯合变异并明确了变异的致病性,拓展了CRPPA基因和MDDG疾病的遗传变异谱和临床表型谱,总结了其临床病例报道和治疗研究,有助于提高对该病的诊断和治疗水平。
