手性冠醚高效液相色谱固定相研究
2023-07-04苏海涛黄韵婷
罗 兰,苏海涛,白 帆,潘 媛,黄韵婷,孔 涛,毛 颖
(云南省产品质量监督检验研究院,云南 昆明 650223)
手性是自然界的基本属性之一[1],广泛存在于化学、物理、医药等各个学科中。随着研究者对手性问题的深入研究,同一对映体的不同构型不仅物理化学性质和光学性质不同,而且在药理学、生理学和毒理学等方面的表现也截然不同,因此手性分离在制药生产、化学工业和生命科学等邻域有重要的研究意义。手性分离的方法有很多,常用分析方法有毛细管电色谱[2]、气相色谱[3-5]、超临界流体色谱[6-7]、高效液相色谱[8-9]等。在手性分离方法中,高效液相色谱是应用最广的方法之一,因其具有高效、高速、应用范围广和灵敏度高等优点。
冠醚是Pederson[10]于1967年首次发现并提出的,它是一种大环聚醚,具有特定尺寸的空腔。分子中的醚氧,作为电子供体配体原子,规律分布在空腔内壁上,金属离子或铵根阳离子引入腔内。手性冠醚是在冠醚分子内引入手性单元,从而使分子具有手性。最早是由Cram将1,1′-二萘基引入到冠醚中,得到联萘型手性冠醚。Cram将该化合物用于手性识别,主要用于去拆分胺类化合物、氨基酸和氨基酯。Shinbo[11]和他的同事也成功合成出R-3,3′-二苯基-1,1′-二萘基-20-冠-6,通过物理方法涂覆在十八烷基硅胶上制备成固定相表现出很好的拆分效果。但国内未见报道R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6的合成及其制备成手性固定相的报道。本文通过以R-联萘酚为原料合成R- (3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6,并将其涂覆于C18硅胶上制备成手性固定相,并对11种对映体得到不同程度的拆分。
1 实验部分
1.1 仪器和试剂
Bruker-500核磁共振仪(Bruker,德国);元素分析仪Vario ELⅢ(德国Elementar公司);ELGALC134净水系统(英国,ELGA);电热鼓风干燥箱(上海,恒科学仪器有限公司);分析天平(梅特勒-托利多仪器有限公司);真空环境在SHZ-D(Ⅲ)循环水式真空泵;EYELA PSL-1810磁力搅拌低温恒温水槽(上海,爱郎仪器有限公司);DF-101S集热式恒温加热磁力搅拌器(予华仪器有限责任公司);高效液相色谱仪(大连依利特分析仪器有限公司);不锈钢色谱柱(250 mm×2.0 mm i.d.)(美国,Alltech)。
R-联萘酚、正丁基锂、三溴化硼(adamas);碘甲烷(西亚);戊乙二醇(Alfa);球形5μm C18硅胶(美国Sepax Technologies);实验测定的手性样品和位置异构体分别购买于Adamas-beta公司、Sigma-Aldrich公司。其余试剂都为国产分析纯。
1.2 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6的合成路线
R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6的合成路线如图1。

图1 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6合成路线
1.2.1 手性2,2′-二甲氧基-1,1′-二萘(B)的合成
搭好装置,检验装置气密性。在氮气保护下,在500 mL圆底烧瓶中加入7 g的R-联萘酚,24 g的无水碳酸钾K2CO3,再向其中加入200 mL的分析纯丙酮,搅拌均匀。在60℃条件下,在恒压漏斗中加入6 mL的碘甲烷缓慢加入反应中。回流10 h,用旋转蒸发仪减压除去反应中的丙酮。残留物用二氯甲烷和蒸馏水萃取3次,再加入适量的无水硫酸镁MgSO4干燥,过滤减压除溶剂。再将所得固体柱纯化,洗脱剂为(V乙酸乙酯∶V石油醚=1∶6),纯化后得白色晶体7.21 g(产率为94%)。1H NMR(500 MHz,CDCl3)δ8.01(d,J=9.0 Hz,2H),7.90(d,J=8.2 Hz,2H),7.49(d,J=9.0 Hz,2H),7.34(ddd,J=8.1,6.7,1.1 Hz,2H),7.24(ddd,J=8.1,6.7,1.2 Hz,2H),7.14(d,J=8.5 Hz,2H),3.79(s,6H)。13C NMR(125 MHz,CDCl3)δ 154.99,134.03,129.40,129.24,127.92,126.30,125.27,123.51,119.64,114.29,56.92。
1.2.2 手性2,2′-二甲氧基-3,3′,6,6′-四溴-1,1′-二萘(C)的合成
称取2 g上一步产物B溶于150 mL的无水乙醚中,在氮气保护下,向其中加入11 mL的液溴,液溴(Br2)在30 min内缓慢滴加完,在室温下搅拌6 h,加入200 mL的无水Na2SO3除去反应中残余的液溴。在用二氯甲烷萃取3次,有机层再用无水Na2SO4除水。减压除去二氯甲烷,所得固体柱纯化,洗脱剂为(V乙酸乙酯∶V石油醚=1∶10)得淡黄色的晶体。R-2,2′-二甲氧基-3,3′,6,6′-四溴-1,1′-二萘(1.6 g,产率40%)。1H NMR(500 MHz,CDCl3)δ8.44(d,J=1.9 Hz,2H),7.78(s,2H),7.34(dd,J=9.0,2.0 Hz,2H),6.95(d,J=9.0 Hz,2H),3.78(s,6H).13C NMR(125 MHz,CDCl3)δ154.75,132.91,130.82,129.47,128.90,127.10,122.79,119.33,119.07,118.34,56.84。
1.2.3 手性R-2,2′-二羟基-3,3′,6,6′-四溴-1,1′-二萘(D)的合成
称取2 g的产物C溶解在200 mL的无水二氯甲烷中,将反应降温到0℃时,先搅拌10 min,缓慢滴加3 mL的三溴化硼溶液,再搅拌25 min后升高温度到室温反应24 h,再将反应降温至0℃,向反应中加入适量的去离子水除去反应中残余的三溴化硼。混合物用二氯甲烷萃取,收集有机相且向其中加入Na2SO4干燥。过滤除去Na2SO4,有机相减压干燥。所得样品需硅胶柱纯化,洗脱剂(V乙酸乙酯∶V石油醚=1∶8),减压除去洗脱剂得浅黄色固体(1.7 g,产率88.9%)。1H NMR(500 MHz,CDCl3)δ8.49(d,J=1.9 Hz,2H),7.77(s,2H),7.45(dd,J=8.9,2.0 Hz,2H),6.98(d,J=8.9 Hz,2H),5.05(s,2H)。13C NMR(125 MHz,CDCl3)δ152.56,132.41,131.94,130.12,129.42,126.17,124.84,123.07,119.90,110.36。
1.2.4 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6(E)的合成
称取2 g的产物D和2.5 g的五甘醇对甲苯磺酸酯、0.24 g的氢氧化钾放入事先搭好的装置中,且保持整个反应环境都是在氮气保护下进行。加入100 mL的无水四氢呋喃到圆底烧瓶中,在65℃下回流72 h,反应完成后用薄层色谱检测。用二氯甲烷和蒸馏水萃取至少三次,有机相收集到大烧杯中并加入无水Na2SO4干燥。硅胶柱纯化,洗脱剂(V乙酸乙酯∶V石油醚=1∶5),除去溶剂得淡黄色粘稠固体(1.24 g,产率为46%)。1H NMR(500 MHz,CDCl3)δ8.33(d,J=1.9 Hz,2H),7.78(s,2H),7.24(dd,J=9.0,2.0 Hz,2H),6.89(d,J=9.0 Hz,2H),4.12-4.08(m,2H),4.01-3.98(m,2H),3.59-3.54(m,4H),3.47-3.40(m,8H),3.34-3.31(m,2H),3.31-3.27(m,2H)。13C NMR(125 MHz,CDCl3)δ 154.45,132.94,130.70,129.38,129.08,127.35,122.37,121.26,119.44,119.35,70.82,70.74,70.60,70.19,69.78。
1.2.5 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性固定相的制作
将0.2 g R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6溶解于8 mL的二氯甲烷中,称取2 g粒径为5μm的C18胶硅置于25 mL圆底烧瓶中,在C18硅胶表面用物理涂敷方法涂敷上述冠醚溶液,旋转蒸发除去有机溶剂,得到R- (3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性固定相。
1.2.6 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6色谱柱的填充
取上述R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性固定相1.4 g和25 mL甲醇/水(体积比)=1/9溶液搅拌,形成悬浮状态后过筛待用,用同体积比的甲醇/水混合溶液作顶替液,40 MPa压力下制备手性色谱柱。
1.2.7 色谱条件
pH=2的高氯酸为流动相;流速0.1 mL/min,柱温25℃。流动相在使用前需经过0.45μm的滤膜过滤,超声除去气泡。
2 结果及讨论
2.1 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6固定相的红外表征
对R- (3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6固定相进行红外光谱表征,图2中A为R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性冠醚;B为固定相;C为C18硅胶。图2结果表明:B与A比较,B中3516 cm-1的峰归属于硅胶表面残存的硅羟基伸缩振动吸收,大约在2926 cm-1和2856 cm-1的峰为手性冠醚中亚甲基的C-H伸缩振动,1582 cm-1为冠醚中芳烃的 =C C伸缩振动。B与C相比较,B在1316 cm-1为冠醚中的部分醚键,在1101 cm-1处的峰信号增强,它为手性冠醚中的C—O—C伸缩振动和C18硅胶本身的Si—O—Si的振动吸收峰,809 cm-1峰为Si—O键对称伸缩振动峰,以上信息可推测硅胶表面成功涂覆上R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6。
2.2 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6的元素分析
对手性固定相进行了元素分析,如表1。

表1 元素分析
元素分析表明,固定相与C18硅胶相比较,碳含量、氢含量有明显的增加,且计算得到实际涂覆量为9.09%,说明R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6已成功涂覆于硅胶表面。
2.3 对外消旋体的拆分
为考察R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6对外消旋体的手性拆分能力。在色谱条件:pH=2的高氯酸流动相(其使用前先用0.45μm滤膜过滤,超声脱气),V(流速)=0.1 mL/min,色谱柱温为T=25℃,色谱柱拆分手性样品检测波长为254 nm,拆分氨基酸波长为210 nm。其中7种手性药物和4种氨基酸得到拆分。它们的结构如图3所示。
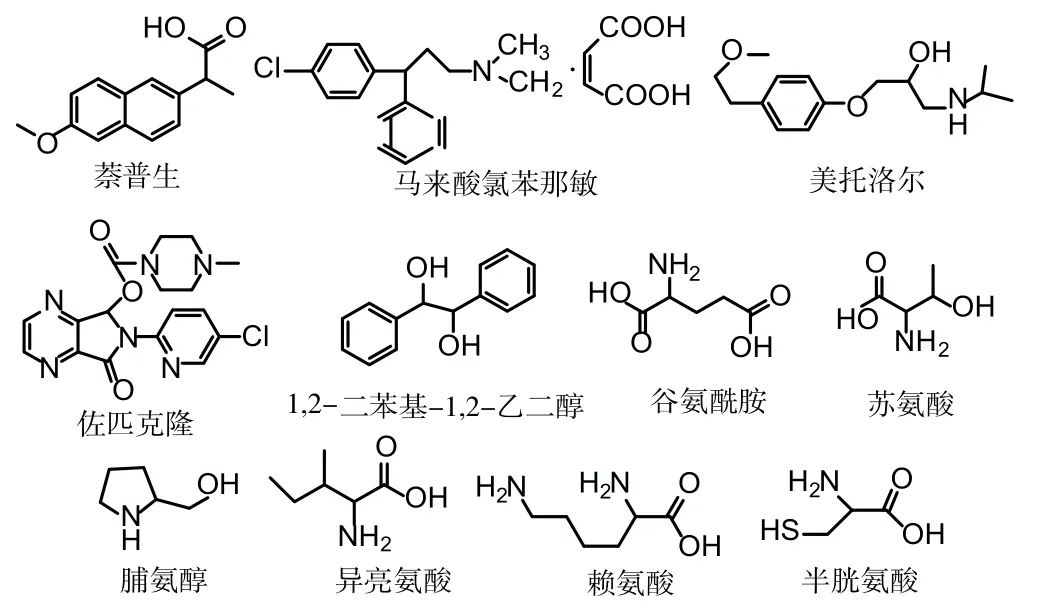
图3 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6色谱柱得到拆分的对映体结构
R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6色谱柱对上述外消旋化合物拆分的结果,如表2所示。
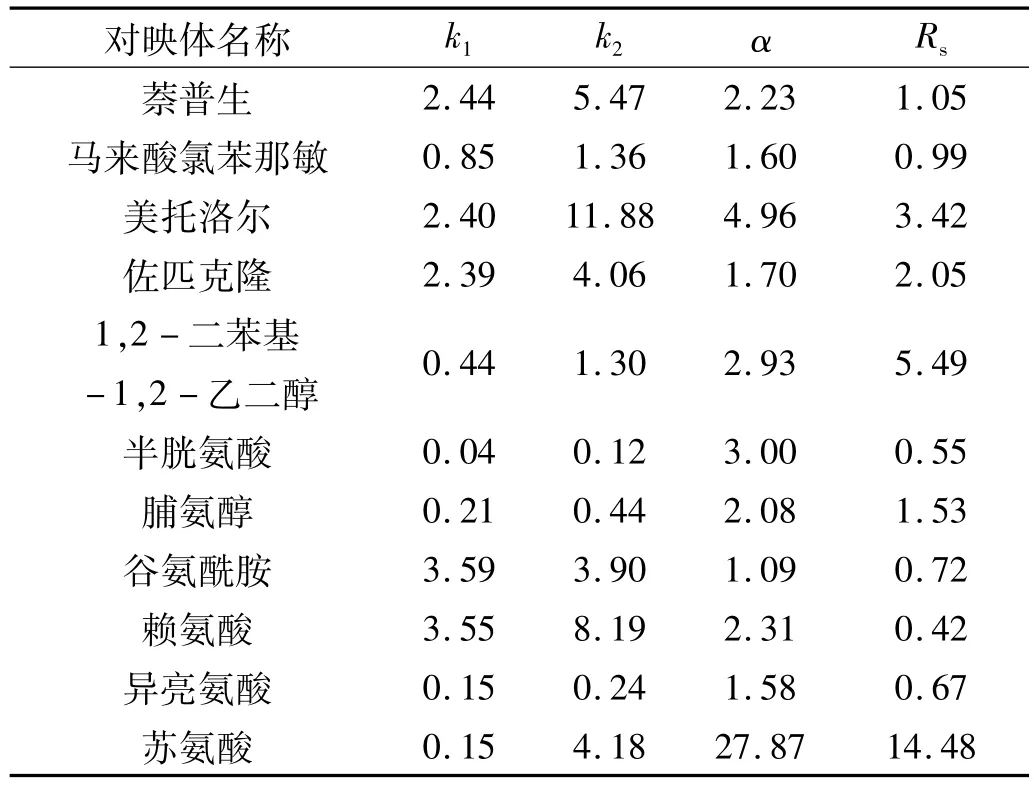
表2 R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性柱对外消旋体的拆分结果
表2中依次列出了保留因子(k1,k2),分离因子(α),和分离度用Rs表示。从表2得出,在R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性冠醚柱中有11种对映体能得到分离。1,2-二苯基-1,2-乙二醇、美托洛尔、佐匹克隆、苏氨酸等外消旋体得到基线分离,萘普生、马来酸氯苯那敏、谷氨酰胺、赖氨酸、异亮氨酸、苏氨酸得到了部分分离,其拆分谱图如图4所示。

图4 手性冠醚色谱柱的色谱分离谱图
2.3.1 手性色谱柱对外消旋化合物的拆分机理
在酸性的环境中,分子中含有伯胺基团的物质和H+形成铵离子,且其极性较强进入冠醚的空腔中作用。在联萘酚的2,3,4,5,6-位置上的取代基对冠醚的手性识别能力有一定的影响,取代基种类影响铵离子在固定相与流动相之间的平衡,从而产生不同的手性识别能力。根据被拆分的化合物的结构,它们基本都含有伯胺基、羟基、羧基等基团。被拆分化合物的基团可能与冠醚分子的氧原子形成氢键,从而达到手性识别。此外,范德华力、色散力、静电作用等作用力也可能对化合物的手性拆分有一定的贡献作用。
2.3.2 色谱柱重现性
为了讨论R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性色谱柱分离手性样品和位置异构体的重现性,在流动相为pH=2条件下,选用萘普生探究柱子的重现性和稳定性,在相同条件下对萘普生重复多次进样后,得到的色谱图如图5。

图5 萘普生重复进样的色谱图
3 结论
本文在手性源为R-联萘酚的基础上,合成了R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6,用物理的方法涂敷在粒径为5μm的C18硅胶上制成R-(3,3′,6,6′-四溴-1,1′-二萘基)-20-冠-6手性冠醚色谱柱。对11种手性化合物得到了拆分,说明该手性色谱柱有一定的手性识别能力。实验表明,手性冠醚用于高效液相色谱固定相较传统的固定相主要拆分氨基酸,该手性柱对几种手性药物也可拆分,拓宽了它的拆分范围。
