Si掺杂调控15-冠-5配位Li+机理的理论研究
2021-11-18梁苏卓成姬国勋孙新利李国东张仕通
梁苏卓成 姬国勋*, 孙新利 李国东 张仕通
(1火箭军工程大学,西安 710025)
(2苏州大学放射医学与辐射防护国家重点实验室,放射医学及交叉学科研究院,江苏省高校放射医学协同创新中心,苏州 215123)
冠醚是一种拥有环状结构和纳米级孔穴的大环聚醚化合物,其化学通式为(CH2CH2O)n。根据环内C和O的个数,常见的冠醚有12⁃冠⁃4、15⁃冠⁃5和18⁃冠⁃6等。由于冠醚的外部骨架表现为疏水性质,而环内的氧原子往往具有很强的负电性,因此该类化合物对金属离子的选择性配位能力较强,在离子识别、同位素分离和催化反应等领域得到了广泛的应用[1⁃3]。此外,对冠醚进行原子掺杂、引入官能团、连接功能性侧链等结构修饰能够改变其物理化学性质,可以进一步拓宽其应用领域。例如,田欢等[4]研究了4,13⁃二硫杂苯并⁃18⁃冠⁃6对软酸Ag+的选择性配位能力。结果表明在多种离子(Cu2+、Pb2+、Zn2+、Ni2+)共存的复杂体系中,4,13⁃二硫杂苯并⁃18⁃冠⁃6对Ag+的萃取效率高达95%。缪谦等[5]以单体1,4⁃二溴⁃2,3⁃萘⁃18⁃冠⁃6和1,4⁃二乙烯基⁃2,5⁃二丁氧基苯通过Pd催化的Heck偶联反应合成了一种能够发射蓝绿色荧光的共轭高分子。通过荧光和紫外-可见光谱测试,他们发现这种共轭高分子能够有效识别Hg2+。
Si与C为同主族的元素,其原子半径大于C,电负性小于C,因此通过Si掺杂的方式能够有效地调控冠醚的骨架及其电子结构性质。在相当长的一段时期内,Si杂冠醚配位金属离子的能力被认为弱于未掺杂的冠醚[6]。这一结论的局限性在于其仅考虑了利用—SiMe2—单元取代—CH2—CH2—的掺杂方式,这种Si掺杂的方法会减少环上的原子数,形成缩环冠醚;而无论是否含有Si原子,缩环冠醚配位金属离子的能力均不如原始的冠醚[7]。近年来,von Hänisch 及 其 合 作 者[8⁃11]报 道 了 一 系 列 含 有—SiMe2—SiMe2—单元的Si杂冠醚,并通过理论结合实验的方法研究了该类结构对金属离子的配位能力。结果表明,Si对冠醚和冠醚-离子配合物的结构有明显的影响,部分Si杂冠醚配位金属离子的能力比未掺杂的冠醚更强。
尽管von Hänisch及其合作者的工作为调控冠醚的金属离子配位性能提供了新的思路,但目前关于通过掺杂—SiMe2—SiMe2—单元调控冠醚金属离子配位性能的研究仍未能系统地揭示其中的调控规律和内在机理。例如,掺杂数量及相对位置对冠醚配位金属离子性能的影响机制尚未被深入理解;对—SiMe2—SiMe2—单元掺杂的冠醚与金属离子间相互作用机理的认识还不够清晰,导致对Si杂冠醚配位金属离子的功能调控和结构设计缺乏有效的理论指导原则。锂在电池、冶金及医疗行业中应用广泛,因此针对锂设计与开发性能良好的冠醚类配位剂具有一定的科学意义及工程价值[12]。鉴于此,我们以15⁃冠⁃5配位碱金属离子Li+为研究模型,利用密度泛函理论(DFT)计算—SiMe2—SiMe2—单元的掺杂数量(1~5组)和掺杂位置对冠醚结构的影响规律,并深入探讨了—SiMe2—SiMe2—单元影响冠醚⁃Li+相互作用的机理。另外,考虑到Si掺杂结构存在更多的可能性,我们还研究了掺杂1组—CH2—SiMe2—单元的结构,作为对—CH2—SiMe2—掺杂方式的初步探索。
1 模型与计算方法
15⁃冠⁃5的环上含有5组—CH2—CH2—单元。我们充分考虑了1~5组—SiMe2—SiMe2—单元掺杂取代—CH2—CH2—单元的情况。掺杂2组和3组—SiMe2—SiMe2—单元时会分别产生2种构造异构体,我们也进行了相应的讨论。另外,虽然以—CH2—SiMe2—单元取代—CH2—CH2—单元的掺杂方式在冠醚的修饰中还未见报道,但是研究者们已经能够合成种类丰富的、含有—CH2—SiMe2—O—单元的稳定环状化合物[13⁃14],说明Si杂冠醚的结构存在更多的可能性。因此,作为理论研究,我们还构建了含有1组—CH2—SiMe2—单元的结构。综上所述,本工作共研究9种冠醚(即15⁃冠⁃5,下同)的结构式,包含1种未掺杂的冠醚和8种Si杂冠醚结构。
为了得到准确的分子构象,我们使用Gaussian 09程序,结合B3LYP泛函[15]和Grimme开发的DFT⁃D3(BJ)色散矫正方法[16],选择 2⁃zeta 基组 Def2⁃SVP[17⁃18]进行了冠醚和冠醚⁃Li+配合物的分子几何结构优化和振动频率计算,并在默认温度298.15 K下计算了吉布斯自由能的热力学校正值。每个优化所得的结构均没有虚频以保证得到的结构对应势能面上的稳定点。基于稳定的波函数和自然布居分析(nat⁃ural population analysis,NPA)计算了原子电荷[19],并使用Multiwfn程序[20]基于分子中的原子理论(atoms in molecules,AIM)进行了电子密度拓扑分析[21]。为了确保能量计算的精度,使用3⁃zeta基组Def2⁃TZVP[17⁃18]进行单点能计算,并结合 counterpoise 方法[22]矫正了基组重叠误差。
气相条件下的冠醚⁃Li+配位反应的吉布斯自由能变(ΔGgas,kJ·mol-1)计算基于式1进行:
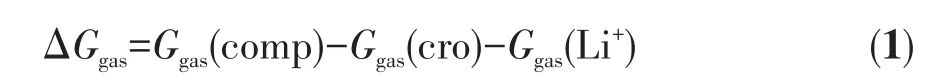
其中Ggas(comp)、Ggas(cro)和Ggas(Li+)分别表示气相条件下冠醚⁃Li+配合物、自由冠醚和Li+的吉布斯自由能。溶剂条件下的冠醚⁃Li+配位反应的吉布斯自由能变(ΔGsol,kJ·mol-1)计算基于式2进行:
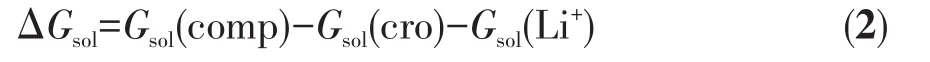
其中Gsol表示相应的物质在溶剂条件下的吉布斯自由能(kJ·mol-1),其计算基于式3。溶剂条件的等效使用了SMD隐式溶剂模型[23]。

值得注意的是,在B3LYP优化结构的基础上,此处气相条件下的单点能Egas和溶剂模型下的单点能Esol计算级别为 M05⁃2X/6⁃31G(d)[24⁃26],这一策略可以更为准确地描述构象在溶剂中的自由能,因此在相关领域的研究中被广泛采用[27]。
冠醚的几何形变能(Egeom)表示冠醚从自由状态下的构型形变为配合物中的构型所需要消耗的能量。Egeom的计算基于式4进行:

其中E(crodis)和E(cro)分别代表形变后的和自由的冠醚的单点能。
冠醚⁃Li+的相互作用能(Eint)的计算基于式5进行:
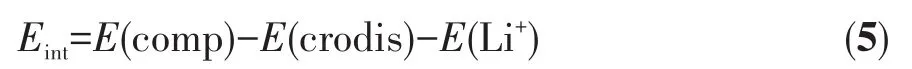
其中E(comp)和E(Li+)分别表示冠醚⁃Li+配合物和Li+的单点能。
为了更加深入地理解冠醚和Li+之间相互作用的类型,本工作进一步将Eint按式6分解:

其中,等式右侧Eelst、Eind、Eexch和Edisp分别代表静电相互作用、诱导相互作用、交换互斥作用和色散作用。各相互作用成分的占比通过PSI4程序[28]中sSAPT0(scaled SAPT0)级别的对称匹配微扰理论分析[29],结合 Jun⁃cc⁃pVDZ基组[30⁃35]得到。根据 Parker等[36]的工作,sSAPT0/Jun⁃cc⁃pVDZ级别的对称匹配微扰理论分析能够可靠地描述分子间各相互作用成分的占比,且计算效率较高,适用于本工作中9个冠醚⁃Li+配合物体系的计算。
2 结果与讨论
由于冠醚骨架的柔性较大,其势能面上可能存在多个极小值点,因此我们对每个结构都从5个初始构象出发进行优化,而后对比所得构象在二氯甲烷(DCM)溶剂中的吉布斯自由能,筛选出吉布斯自由能最低的构象作为相应分子的最优构象。对于自由冠醚(L),我们根据文献[37]报道的 15⁃冠⁃5冠醚结构建立1个初始冠醚构象,并在此基础上创建4种冠醚⁃M(M=Na+、K+、Mg2+、Ca2+)配合物模型(初始设置M在5个O形成的平面中心,且与5个O距离相同)。对这4种配合物模型进行结构优化计算,将优化后构象中的金属离子去除,得到另外4种冠醚初始构象。在这5种初始的冠醚构象的基础上,取代碳原子并建立相应的Si掺杂模型。对于每种类型的冠醚⁃Li+配合物,我们将Li+分别放置在5个冠醚初始构象的环中5个O形成的平面中心,且与5个O距离相同,得到5个冠醚⁃Li+初构象。而后优化并计算所得构象在DCM溶剂中的吉布斯自由能,筛选出吉布斯自由能最低的构象。图1所示为筛选得到的配位Li+前后冠醚的最稳定构象。
从图中可以发现,Li+均稳定地配位在冠醚环内位置,且造成冠醚的结构发生了明显变化。为了更加清晰地认识冠醚的形变程度,我们定量地统计了配位前后冠醚结构中的键长(包含C—C、Si—Si、C—O 和 Si—O)、键角(如 C—O—C、Si—O—Si以及 O—C—O)等结构特征的平均值。如表1所示,配位前后冠醚环上的 C—C、Si—Si、C—O、Si—O 键长和 C—O—C、C—O—Si键角仅发生了小幅度的变化,而Si—O—Si的变化较为明显,说明冠醚的形变主要依靠σ键的旋转和Si—O—Si键角的变化来进行。这里值得注意的是,配位Li+之后,冠醚的形变一方面需要吸收能量,是配合物稳定性的不利因素;另一方面可形成良好的冠醚⁃Li+配位结构,是配合物稳定性的有利因素。即配合物的结构受到冠醚本身的形变能以及冠醚⁃Li+之间相互作用能的共同影响,因此不同冠醚配位Li+产生的形变程度不同。
通过图1可以看出L⁃Li+中的5个O和Li+几乎分布在同一个平面内,并且Li+位于环状结构中心位置,主要与环内侧的O配位。由于Si—Si、Si—O键比C—C、C—O键更长,键角C—O—Si和Si—O—Si的角度比C—O—C更大,因此掺杂Si会增大冠醚环的尺寸,导致掺杂 1 组—SiMe2—SiMe2—的 LSi⁃Li+、掺杂 1 组 —CH2—SiMe2— 的 LSiC⁃Li+和 掺 杂 2 组—SiMe2—SiMe2—的 L2Si⁃O⁃Li+、L2Si⁃M⁃Li+中冠醚环产生了扭曲。但是5个O均能与Li+靠近形成配位,且整体上O—Li+键长较L⁃Li+有所缩短(表1)。继续增加Si的数量,由于冠醚的尺寸进一步增加,因此L3Si⁃O⁃Li+、L3Si⁃OM⁃Li+和 L4Si⁃Li+中的 O3 都明显远离 Li+。在所有—CH2—CH2—都被—SiMe2—SiMe2—取代的 L5Si⁃Li+中,O3和O5远离Li+,剩余的3个O与Li+成3配位结构(图 1)。

表1 自由冠醚及冠醚-Li+配合物的几何结构参数Table 1 Geometrical parameters of free crown ethers and crown-Li+complexesa
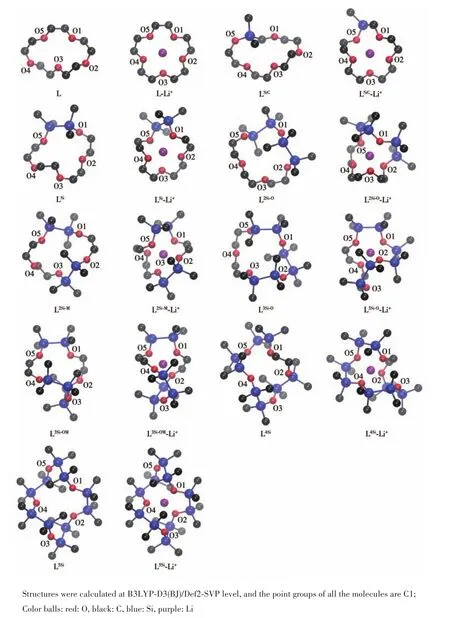
图1 DFT优化的15⁃冠⁃5配位前后的几何结构Fig.1 DFT optimized geometric structures of 15⁃crown⁃5 before and after coordination
总之,9种冠醚⁃Li+配合物中的O—Li+配位键长均在0.191~0.225 nm范围,与实验得到的Li(1,1,2,2,4,4,5,5⁃8甲基⁃1,2,4,5⁃4硅杂⁃10,11⁃苯并⁃15⁃冠⁃5)I晶体中O—Li+配位键长(0.201 6~0.227 6 nm)[10]相近,明显大于O的原子半径0.066 nm[38]与Li+的离子半径0.059 nm[39]之和,因此从键长数据来看,O—Li+形成的是弱配位键。
由于冠醚对Li+的配位常在DCM溶剂中进行,且von Hänisch等[8⁃11]的工作表明DCM也可以作为Si杂冠醚的溶剂。因此为了比较9种冠醚配位Li+的能力,我们分别计算了气相条件下和DCM溶剂条件下冠醚⁃Li+配位反应的吉布斯自由能变,结果如图2所示。从图 2 可以发现,气相条件下,LSi、L2Si⁃M、L3Si⁃OM、L4Si配位 Li+的能力在热力学上比 L 更具优势,L2Si⁃O配位 Li+的能力与 L 相差不大,LSiC、L3Si⁃O和 L5Si配位Li+的能力比L更弱,说明掺杂Si能够在热力学上有效调控冠醚配位Li+的能力,调控的效果受掺杂数量、掺杂相对位置的影响。在DCM溶剂条件下的冠醚⁃Li+配位反应在热力学上的可行性被明显削弱,这主要是由于在溶剂中Li+会与溶剂分子形成团簇,而冠醚配位Li+导致一部分溶剂分子被脱除,这个过程会消耗能量[40]。通过对比气相条件下和DCM溶剂条件下的数据可知,9种冠醚与Li+的配位反应的热力学可行性被削弱的程度不同,并且整体上Si杂冠醚被削弱得更明显。在DCM溶剂条件下,仅L3Si⁃OM对 Li+的配位强于L,相应配位吉布斯能比 L⁃Li+低4.60 kJ·mol-1。据报道,von Hänisch及其合作者[9]通过实验观察到在DCM溶剂中,通过Si掺杂12⁃冠⁃4形成1,1,2,2⁃4甲基⁃1,2⁃2硅杂⁃12⁃冠⁃4,也可以增强冠醚对Li+的配位能力。这些结果说明通过掺杂Si能够有效调控冠醚在气相和溶剂条件下配位金属阳离子的能力。
为了进一步揭示冠醚中O—Li+的成键本质,以及Si影响冠醚⁃Li+相互作用的机理,我们对9种冠醚⁃Li+配合物进行了电子密度AIM拓扑分析(表2)。表2的数据显示,每一个相互靠近形成配位的O—Li+之间都出现了键临界点(BCP),而相互远离的O—Li+之间没有BCP。BCP上的电子密度(ρ)、电子密度的拉普拉斯函数(∇2ρ)、电子能量密度(H)和电子势能密度绝对值(|V|)与动能密度(K)的比值(|V|/K)能够用于判断成键原子之间的相互作用类型。对于共价作用,原子间存在定域性强的共用电子对,BCP处通常对应∇2ρ<0、H<0、|V|/K>2,且ρ>0.14 a.u.的性质[41⁃43]。而对于闭壳层作用(非共价作用),原子间没有共用电子对,BCP处通常具有∇2ρ>0、H>0、|V|/K<1,且ρ<0.14 a.u.的性质。通过表2可以看出,所有O—Li+的BCP处的ρ都很小(不超过0.04 a.u.),且∇2ρ>0、H>0、|V|/K<1,具有明显的非共价作用特征,即Li+对O电子的极化效应较弱,不足以使O与Li+之间形成共用电子对。因此,9种冠醚与Li+形成的O—Li+配位键本质上是闭壳层相互作用。

表2 O—Li+配位键BCP处拓扑电子密度性质Table 2 Topological electron density properties at BCP of O—Li+coordination bond*

续表2
为了深入理解掺杂Si对冠醚与Li+之间的相互作用强度的影响,我们在B3LYP⁃D3(BJ)/Def2⁃TZVP级别计算了冠醚⁃Li+的相互作用能以及冠醚的形变能,并在sSAPT0/Jun⁃cc⁃pVDZ级别进行了相互作用能能量分解分析以研究其中各相互作用成分的占比及变化,数据展示在表3中。其中,Eelst描述冠醚⁃Li+配合物中,冠醚与Li+的电子密度互不影响的情况下相互之间的静电作用;Eind通常用来描述轨道极化和电子转移产生的诱导相互作用;Eexch用于描述冠醚与Li+之间,由于占据态轨道间Pauli排斥作用导致的配合物整体能量升高;Edisp用来描述冠醚与Li+之间的色散相互作用。
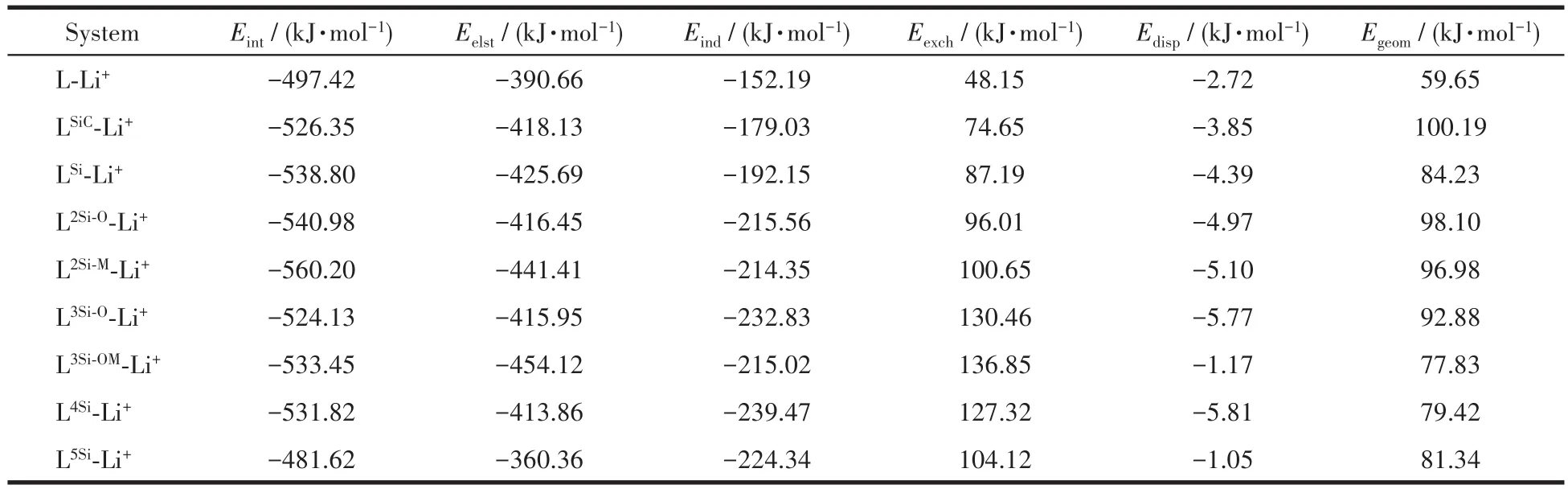
表3 冠醚-Li+相互作用能及能量分解Table 3 Crown-Li+interaction energies and energy decompositions*
结果显示,2种级别下计算得到的冠醚⁃Li+相互作用能大小基本一致。掺杂1组—CH2—SiMe2—的LSiC与Li+的相互作用比L更强(ΔEint=28.93 kJ·mol-1),掺杂1组—SiMe2—SiMe2—的LSi与Li+的相互作用比L 更 强 (ΔEint=41.38 kJ·mol-1),表 明 以 —SiMe2—SiMe2—单元或—CH2—SiMe2—单元取代—CH2—CH2—单元能够增强冠醚与Li+的相互作用。掺杂2组—SiMe2—SiMe2—的 L2Si⁃M与 Li+的相互作用强度得到了进一步的提升,比掺杂1组—SiMe2—SiMe2—的LSi更强(ΔEint=21.40 kJ·mol-1)。然而,L2Si⁃O与 Li+的相互作用强度较LSi并没有明显的优势,证明当掺杂多组—SiMe2—SiMe2—时,掺杂的相对位置对冠醚结合Li+的能力有一定影响。继续掺杂更多的—SiMe2—SiMe2—会导致Li+配位数减少。尽管如此,与Li+形成 4 配位的 L3Si⁃O、L3Si⁃OM和 L4Si与 Li+的相互作用依旧比L更强。L5Si由于仅能与Li+形成3配位结构,与Li+的相互作用强度比L更弱。另外,数据显示掺杂—SiMe2—SiMe2—或—CH2—SiMe2—也会增加冠醚的形变能,扣除形变能后 LSi、L2Si⁃O、L2Si⁃M、L3Si⁃OM和L4Si的Eint较L分别有 16.80、5.10、25.46、17.85、14.63 kJ·mol-1的优势。
能量分解结果表明,冠醚⁃Li+相互作用中静电相互作用的占比最大,诱导相互作用的占比较低,色散作用仅起到微弱的贡献。结合电子密度AIM拓扑分析的结果,可以证明冠醚与Li+的相互作用本质是伴随少量轨道极化和电子转移的离子-偶极相互作用。掺杂Si对静电相互作用和诱导相互作用有一定增强效果,交换互斥作用也随着Si数量的增加而增强。另外,由于Si的引入,色散相互作用也得到了略微增强,而 L3Si⁃OM⁃Li+和 L5Si⁃Li+中色散相互作用较弱的原因是 L3Si⁃OM⁃Li+中的 O3,L5Si⁃Li+中的 O3和O5与邻近的Si一起远离Li+。诱导相互作用的增强一方面来源于掺杂Si导致的O—Li+距离缩短,另一方面源于Si的电子比C更易被极化。交换互斥作用的增强主要是由于O与Li+距离缩短,以及冠醚环发生扭曲导致的Li+在整体上与冠醚框架距离更近。掺杂—SiMe2—SiMe2—的相对位置对冠醚⁃Li+相互作用的影响主要体现在静电相互作用项。
我们对9个冠醚⁃Li+体系进行了NPA以进一步研究掺杂Si调控冠醚⁃Li+静电相互作用的机理,结果列在表4中。数据显示,冠醚中的O带有负电荷,并且与Si相邻的O带有明显更多的负电荷,说明电负性低的Si向相邻的O转移了更多的电子,从而使O与Li+产生了更强的静电吸引。而L2Si⁃M⁃Li+和 L3Si⁃OM⁃Li+中 5 个 O 的 电 荷 总 量 大 于 L2Si⁃O⁃Li+和L3Si⁃O⁃Li+,说明当掺杂 Si的数量相同时,使更多的 O与Si相邻能够让O更加充分地从Si获得电子。此外,由于C几乎不带电荷,而Si带有正电荷,说明掺杂Si后,Si与Li+之间的静电互斥会在一定程度上抵消O与Li+之间的静电吸引作用,这个现象也存在于—SiMe2—取代—CH2—CH2—的冠醚中,并导致了这类冠醚配位金属离子的能力被削弱[44]。而键角Si—O—Si大于C—O—Si表明Si—O—Si单元不利于Si远离Li+,因此会导致Si与Li+之间的静电互斥作用增强。
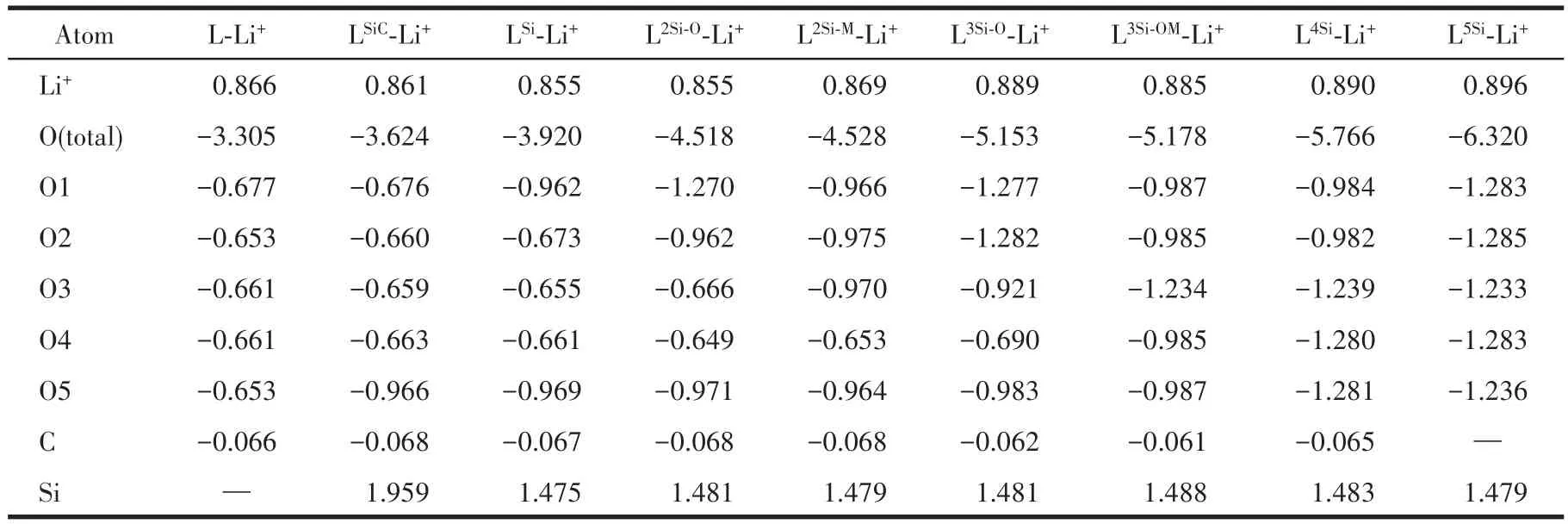
表4 冠醚-Li+配合物的NPATable 4 NPA of crown-Li+complex*
以上分析表明,Si向O转移了大量电子是增强冠醚⁃Li+静电相互作用的关键因素。而当掺杂Si的数量相同时,使更多的O与Si相邻,减少Si—O—Si单元,能在整体上使O获得更多电子,并使Si更加远离Li+,从而增强冠醚结合Li+的能力。因此根据本工作中的计算结果,L2Si⁃M和 L3Si⁃OM与 Li+的相互作用比 L2Si⁃O和 L3Si⁃O更强。
3 结 论
运用密度泛函理论深入地研究了掺杂Si对15⁃冠⁃5配位Li+的影响。结果表明,掺杂—SiMe2—SiMe2—或—CH2—SiMe2—能够增大冠醚环的尺寸,导致掺杂3组及以上—SiMe2—SiMe2—单元会降低冠醚中O与Li+的配位数。通过气相条件和DCM溶剂条件下的冠醚⁃Li+配位反应吉布斯自由能变计算发现:从热力学角度出发,气相条件下 LSi、L2Si⁃M、L3Si⁃OM、L4Si配位 Li+的能力比 L 更强,L2Si⁃O配位 Li+的能力与L相差不大。DCM溶剂条件会削弱配位反应的热力学可行性(相应吉布斯自由能变升高),且整体上Si杂冠醚被削弱的程度更高。但是DCM中L3Si⁃OM在热力学上配位Li+的能力依旧强于L。根据冠醚⁃Li+配合物中的电子密度AIM拓扑分析结果,以及冠醚⁃Li+相互作用能能量分解结果,我们发现本工作所研究的9种冠醚与Li+的相互作用本质均为伴随少量轨道极化和电子转移的离子-偶极相互作用。掺杂Si能够增强冠醚与Li+之间的静电相互作用和诱导相互作用,但是冠醚⁃Li+配合物中的冠醚环发生扭曲会增加冠醚与Li+之间的交换互斥作用。NPA表明,电负性弱的Si向相邻的O转移了大量的电子是—SiMe2—SiMe2—或—CH2—SiMe2—增强冠醚与Li+之间的静电相互作用的关键因素,但同时带正电的Si与Li+产生的静电排斥会在一定程度上抵消O对Li+静电吸引的优势。因此为了提高Si杂冠醚配位Li+的能力,需要尽量避免出现Si—O—Si结构(键角较大,会导致 Si与 Li+的距离缩短),从而使Si充分远离Li+。最后,结合以上的分析,我们认为要得到配位金属离子能力较强的Si杂冠醚,其分子结构设计需要遵循以下2个规则:一是使冠醚环的尺寸与目标离子相匹配;二是尽量避免冠醚环内出现Si—O—Si单元。
本工作从理论机理层面验证了掺杂—SiMe2—SiMe2—对冠醚配位金属离子能力的调控机理,同时对掺杂—CH2—SiMe2—的冠醚结构进行了探索,并提出了Si杂冠醚分子结构设计需要遵循的规则,对通过掺杂Si进行冠醚的结构修饰,以及有机Si化合物与金属离子的相互作用研究提供了一定的理论依据及研究思路。
猜你喜欢
杂志排行

无机化学学报的其它文章
- Mn-Based Coordination Polymer:Facile Synthesis,Structure and Application in Glucose Electrochemical Sensing
- Preparation of Dehydrated Ni-Fe Hydrotalcite-like Compounds as an Eco-Friendly Catalyst for Highly Selective Acetalization of Biomass-Derived Furfural
- Synthesis,Structure Regulation and Characterization of Cadmium(Ⅱ)Complexes Based on Imidazole Carboxylic Acid Ligands
- Difunctional Effects of Organo-Modified T-Type Zeolite Membranes for Dewatering from Organic Solution
- Controlling Distribution of Gold Nanoparticles in Au@ZIF-8 Core-Shell Structures for Sensing Fluorescent Molecules with Photoluminescence
- Structures and Luminescence Property of Two Co(Ⅱ) and Cd(Ⅱ)Supramolecular Coordination Networks Created via Synergistic Effect of Coordination Bonds and Secondary Interactions