高效液相色谱法测定水中α-萘酚和β-萘酚
2016-11-29张国祯李丹丹马可婧
张国祯,李丹丹,马可婧
(甘肃省环境监测中心站,甘肃 兰州730020)
高效液相色谱法测定水中α-萘酚和β-萘酚
张国祯,李丹丹,马可婧
(甘肃省环境监测中心站,甘肃 兰州730020)
建立了液液萃取—高效液相色谱法测定水样中α-萘酚和β-萘酚的分析方法。对流动相组成及流速、柱温、萃取剂种类、色谱柱类型等条件进行了优化。选用三氯甲烷为萃取剂,乙腈:水(0.1%乙酸)=50:50为流动相,流速1mL/min,柱温40℃,作为色谱条件。在1L空白水样中添加低浓度水平的萘酚标准溶液(加标量为0.2μg),测定平行样品7份,α-萘酚加标回收率为92%~117%,标准偏差为5.5%;β-萘酚加标回收率为96%~121%,标准偏差为5.1%。当萃取体积为1L,浓缩至1mL,进样量为10μL时,α-萘酚和β-萘酚的方法检出限分别为0.15μg/L和0.17μg/L。
液液萃取;高效液相色谱;α-萘酚;β-萘酚
萘酚属于多环芳烃类物质的一种,由于羟基位置的不同有两种同分异构体,分别为α-萘酚和β-萘酚,是医药、农药、染料、化妆品生产合成等行业的重要中间体,有毒,对皮肤、黏膜有强烈的刺激作用,易于经皮肤吸收。在我国环境行业,《地表水环境质量标准》(GB3838-2002)、《污水综合排放标准中》(GB8978-1996)以及医药等行业排放标准中,水环境萘酚的各类水质限量标准和标准检验方法尚未建立,目前仍将其归类于挥发酚中。2015年环保部组织征集水体中萘酚类化合物的测定方法。因此,研究水中萘酚类化合物的的测定方法具有重要的意义。
目前,α-萘酚和β-萘酚的测定方法主要有紫外光谱法[1]、荧光光谱法[2-5]、高效液相色谱法[6-8]、电化学分析法和联用技术[9]。关于环境水体中α-萘酚和β-萘酚同时测定的方法报道不多。杨彤通过固相萃取,利用U-BondapackC18(0.39cm×30cm)色谱柱,甲醇和水(含1%乙酸)为流动相,完成了水体中α-萘酚和β-萘酚的同时测定[10]。本文采取液液萃取-高效液相色谱法成功实现了清洁地表水中α-萘酚和β-萘酚的测定。
1 实验材料与方法
1.1 样品采集与保存
采样选用棕色玻璃瓶灌装,采样时水样应充满采样瓶并加盖密封,防止空气进入,样品在4℃下避光保存。
1.2 仪器与试剂
仪器:配有四元梯度泵和荧光检测器(FLD)的液相色谱仪(Aglient1200)、液相色谱柱Discovery@C18 (25cm×4.6mm,5μm)、分液漏斗(1000mL)、氮吹仪(NEVAP 111)、有机相滤头(津隆,有机,0.22μm)。
试剂和水:甲醇、乙腈、二氯甲烷、三氯甲烷、四氯化碳均为色谱纯级,乙酸等均为优级纯,实验用均为超纯水。
标准品:α-萘酚标准品(Accustandard),浓度为100mg/L;β-萘酚标准品,纯度为99.0%。
1.3 标准曲线绘制
称取10mgβ-萘酚,使用甲醇溶解并转移到100mL容量瓶中,定容至刻度线,β-萘酚浓度为100mg/L。将浓度为100mg/L的α-萘酚和β-萘酚标准溶液分别稀释至50mg/L、20mg/L、10mg/L。依次移取 20mg/Lα-萘酚和β-萘酚标准溶液 10、25、50、100μL,定容到1mL,移取50ppmα-萘酚和β-萘酚标准溶液100μL,定容到1mL,配制成浓度分别为0.2、0.5、1、2、5、10mg/L的混合标准溶液,用于绘制外标标准曲线。
1.4 样品前处理
量取1000mL水样到1000mL分液漏斗,加入20mL三氯甲烷,振摇5min,静置分层,萃取3次,合并有机相,有机相采用氮吹仪浓缩至1mL。
1.5 样品分析
进样体积为10μL,流速为1.0mL/min,色谱柱为Discovery@C18(25cm×4.6mm,5μm),柱温为40℃,荧光检测波长为λex=282nm,λem=480nm,流动相为乙腈:水(0.1%乙酸)=50:50。等梯度洗脱,流速为1mL/min。
2 实验结果与讨论
2.1 流动相选择
分别选用甲醇—水、乙腈—水、甲醇—水(0.1%乙酸)、乙腈—水(0.1%乙酸)为流动相,通过比较发现乙腈:水=55:45和 甲醇:水(0.1%乙酸)=55:45条件下,峰面积相对较小,峰宽较大,且色谱系统柱压较高,故选择乙腈:水=50:50和乙腈:水(0.1%乙酸)= 50:50体系。乙腈+水体系下,综合峰高、峰面积、峰宽、响应因子等因素,最终选择乙腈:水(0.1%乙酸)= 50:50,如图1所示。
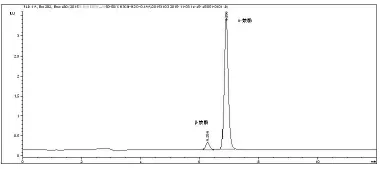
图1 色谱分离谱图
2.2 色谱柱和柱温、流速的选择
选用了不同厂家不同型号色谱柱,通过比较发现液相色谱柱Discovery@C18(25cm×4.6mm,5μm)峰型和峰面积较好。
对不同柱温(25、30、40℃)条件下萘酚类化合物的分离效果进行比较,发现柱温对峰高和峰面积的影响变化不大,但峰宽依次略有降低,所以色谱柱温选择40℃。
比较了 0.8、1、1.2mL/min条件下色谱分离效果,α-萘酚和β-萘酚的分离度依次减小。0.8mL/min保留时间较长,峰宽较大。1.2mL/min两峰的分离度较差,故最终选择流速为1mL/min。
2.3 检测波长的选择
通过参考文献比较,选用荧光检测波长分别为λex=282nm,λem=480nm。
2.4 萃取条件的优化
选用二氯甲烷、氯仿、四氯化碳做为萃取有机溶剂,在空白样品中分别加标2mg/L、10mg/L各100μL,按照样品前处理方法进行萃取。浓缩至1mL,取10μL进入色谱柱,通过比较发现使用氯仿作为萃取溶剂效率较高,回收率较好。见表1。
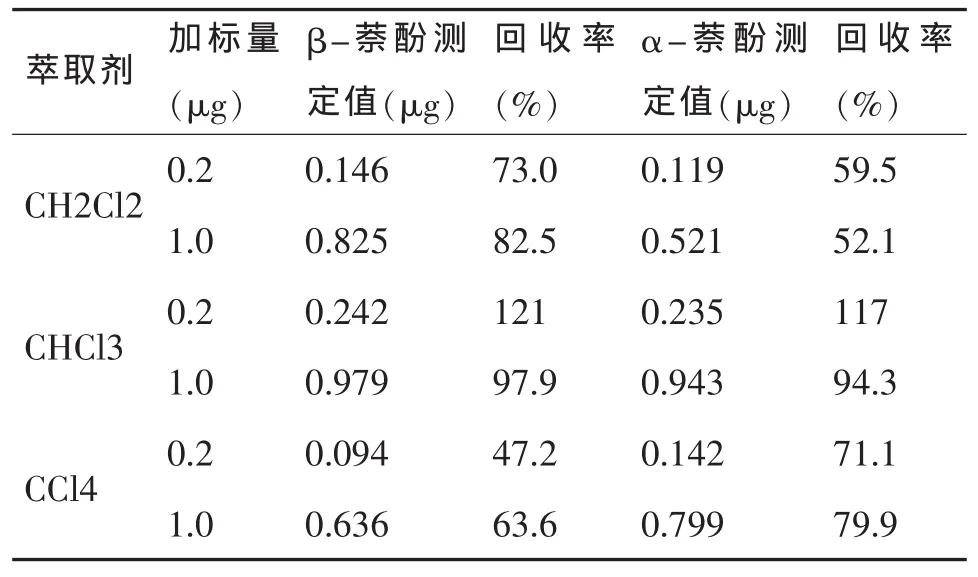
表1 不同萃取条件回收率测定
2.5 线性范围、相关系数和检出限
配制0.2、0.5、1、2、5、10mg/L的萘酚类化合物混合标准溶液,得到α-萘酚和β-萘酚不同浓度响应值,通过线性方程拟合,其标准曲线、相关系数、方法检出限见表2。实验表明,各萘酚浓度为0.2~10mg/L具有良好的线性关系,相关系数>0.9997。

表2 各萘酚化合物标准曲线、相关系数和方法检出限
在1L空白水样中加入低浓度萘酚类化合物(加标量为0.2μg),按照样品分析步骤平行测定7份,根据3倍标准偏差计算两种萘酚类化合物的方法检出限。当萃取体积为1L,浓缩至1ml,进样量为10μL时,α-萘酚和β-萘酚的方法检出限分别为0.15μg/L和0.17μg/L。
2.6 方法准确度和精密度
在1L空白水样中添加低浓度水平的萘酚标准溶液(加标量为0.2μg),平行样品7份,按照上述前处理和样品分析方法,测得加标回收率和相对标准偏差,见表3。
3 结论
以三氯甲烷为萃取剂,乙腈:水(0.1%乙酸)=50: 50为流动相,流速1mL/min,柱温40℃的液液萃取—高效液相色谱法,能够同时测定水体中α-萘酚和β-萘酚,具有良好的线性关系、准确度、精密度。当萃取体积为1L,浓缩至1ml,进样量为10μL时,α-萘酚和β-萘酚的方法检出限分别为0.15μg/L和0.17μg/L。该方法适用于地表水中α-萘酚和β-萘酚的测定,具有一定的应用和推广价值。

表3 α-萘酚和β-萘酚7次空白加标平行分析结果
[1] 刘美君,高国正.双波长紫外分光光度法同时测定-萘酚和-萘酚的含量[J].郑州大学学报:自然科学版,1990,22(2):83-86
[2] 胡敬田,杨景和,周广军,等.荧光同步扫描-双波长标准加入法同时测定-萘酚和-萘酚[J].分析化学,1996(9): 1059-1061.
[3] 李耀群,黄贤智,许金钩,等.导数-可变角同步荧光法用于1-萘酚和2-萘酚的同时测定[J].高等学校化学学报,1993,14(3):334-335.
[4] 巫淼鑫,郑用熙.-环糊精增敏荧光法分别测定1-萘酚和2-萘酚混合物中各自含量[J].江苏石油化工学院学报,1994,6(Z1):42-46.
[5] 周纯,王琼娥,庄惠生,等.直接荧光光度法同时测定水中痕量的1-萘酚2-萘酚[J].光谱学与光谱分析,2008,28(1):2628-2632.
[6] 张孝松,林长山,何天敬,等.反相高效液相色谱法同时测定1-萘酚和2-萘酚[J].化学试剂,1989,11(8):129-131.
[7] 段文胜,干丽君.高效液相色谱法测定1,5-萘二酚中的1-萘酚和2-萘酚[J].上海染料,2003,31(2):31-32.
[8] 黎俊宏,李贵荣,唐宏兵,等.反相高效液相色谱法测定尿中-萘酚-萘酚对硝基酚和间硝基酚[J].光谱实验室,2011,28(2):782-786.
[9] 叶存玲,刘清玲,王治科,等.分散液相微萃取-高效液相色谱法测定水中-萘酚和-萘酚[J].分析试验室,2010,29(8):40-43.
[10]杨彤.高效液相色谱法同时测定水中的-萘酚和-萘酚[J].广州化工,2012,40(7):143-144.
X832
