硅杂原子提升冠醚对锂离子络合能力的机理理论研究
2021-06-30梁苏卓成姬国勋孙新利王波张仕通代星
梁苏卓成,姬国勋,孙新利,王波,张仕通,代星
(1西安高科技研究所,陕西西安710025;2苏州大学放射医学与辐射防护国家重点实验室,放射医学及交叉学科研究院,江苏省高校放射医学协同创新中心,江苏苏州215123;3东北电力大学理学院,吉林省吉林市132012)
引 言
冠醚是一类聚醚分子,一般由—CH2—O—CH2—单元构成,拥有环状柔性骨架结构。根据环内C和O的个数,冠醚被命名为15-冠-5、18-冠-6等,其中第一个数字代表C和O的总个数,第二个数字代表O的个数。冠醚分子整体呈电中性,分子中的O拥有比C和H更高的电负性,因此电子更多地向O聚集,从而能够通过电荷-偶极以及配位共价键等作用络合金属离子,具有对碱金属和碱土金属的强络合能力[1-3]。改变冠醚分子环状骨架的大小、引入N或S等杂原子、增加官能团、连接具有不同功能的侧臂能够调整冠醚对金属离子的络合性能,使其具有对特定金属离子的选择络合性[4-6]。引入具有发光特性的官能团,结合冠醚的强络合能力,研究者获得了多种具有金属离子荧光识别能力的冠醚[7-9]。由于冠醚对金属离子的络合与荧光识别都以冠醚和金属离子的相互作用为基础,因此通过结构设计得到对金属离子作用能力更强的冠醚一直是研究热点。
Si与C在元素周期表中属于同族元素,化学性质具有相似之处,然而相应的Si与C化合物可能表现出截然不同的性质,例如室温下CO2呈气态而SiO2为晶体。近年来,含有Si杂原子的类醚分子与金属离子的相互作用受到了研究者的关注[10-12]。由于Si具有比C更弱的电负性,含有Si杂原子的冠醚中与Si相邻的O会带有更多的负电荷,因此可能与金属离子产生更强的电荷-偶极相互作用与配位共价作用,从而拥有比不含Si的有机醚更强的金属离子络合能力。然而研究表明,由于O与邻近的Si—H、Si—Me反键轨道产生的超共轭作用[13]、带有更多正电荷的Si对金属离子产生的库仑互斥作用,以及更大的形变能[14],Si杂冠醚往往表现出比有机冠醚更弱的金属离子络合能力[15]。对此,Hänisch等[16-18]基于实验与密度泛函理论(DFT)计算结果提出了不同的观点,认为当相邻的O间隔两个—SiMe2—单元时,Si杂冠醚能够比相应的有机冠醚更稳定地络合金属离子,例如1,1,2,2-4甲基-1,2-2硅杂12-冠-4拥有比12-冠-4更强的锂离子络合能力。尽管Hänisch等对Si杂冠醚与金属离子的相互作用研究仅停留在配位键键长、相互作用能和形变能层面,但是足以证明,通过合理引入Si杂原子,可以获得与金属离子相互作用能力更强的冠醚材料,从而进一步提升冠醚类金属离子吸附、荧光识别材料的性能。
目前,Si杂冠醚结构设计所需的理论基础还不够成熟,Si杂冠醚的性质及其与金属离子的相互作用机理仍有待深入理解。因此,本文通过DFT理论计算,详细地探讨了1,1-2甲基-1-硅杂12-冠-4(以下简称LSi/C)、1,1,2,2-4甲基-1,2-2硅杂12-冠-4(以下简称LSi)、1,1,2,2,4,4,5,5-8甲基-1,2,4,5-4硅杂12-冠-4(以下简称LSi-O)、1,1,2,2,7,7,8,8-8甲基-1,2,7,8-4硅杂12-冠-4(以下简称LSi-M)及原始12-冠-4(以下简称L)与Li+的相互作用,并揭示了Si杂冠醚对Li+络合增强的机理。考虑到在实际的工程应用中,冠醚与Li+的络合反应往往在溶剂中进行,而溶剂环境对此类络合反应的Gibbs自由能变有不可忽略的影响[19]。因此,本文还计算了气相条件下和二氯甲烷溶剂条件下5种冠醚-Li+络合反应的Gibbs自由能变,以研究溶剂中5种冠醚对Li+的络合能力,以及有机溶剂二氯甲烷对络合反应的影响。
1 模型与计算方法
基于前人的研究基础,即认为当—SiMe2—SiMe2—单元取代相邻的O间—CH2—CH2—单元时,双Si杂冠醚能够比相应的有机冠醚更稳定地络合金属离子。此掺杂方式不会改变冠醚大环原子数,对于12-冠-4来说,掺杂前后均为12元环。因此本工作采取掺杂Si数量为1的LSi/C、掺杂Si数量为2的LSi、掺杂Si数量为4的LSi-O和LSi-M以及原始未掺杂的12-冠-4共5种冠醚作为理论计算模型(图1),探究5种冠醚对Li+的相互作用。本工作的研究内容同时涉及Si的掺杂数量和掺杂位点对冠醚性质的影响。以—SiMe2—取代—CH2—CH2—的单硅杂冠醚已经被实验确认其络合Li+能力不如原始冠醚[15],主要原因理论上被解释为带正电的Si与Li+产生库仑排斥[11],因此该模型不在本文讨论范围。

图1 冠醚的化学结构式Fig.1 The chemical formulas of crown ethers
由于冠醚-金属离子络合中的相互作用大量涉及电子层面的机理,高精度量子化学计算适合研究此类问题[20-22]。基于计算精度和效率的同时考虑,本文采用Gaussian 09程序对所有体系执行密度泛函理论计算。
在B3LYP-D3(BJ)/Def2SVP级别[23-28]对体系进行几何优化。优化过程中没有对对称性施加任何限制。在相同理论级别下进行了频率计算,没有虚频以保证得到的结构是势能面上真正的驻点。首先优化的结构是12-冠-4。而后在所得的稳定12-冠-4结构上掺杂Si并进行优化,得到4种Si杂12-冠-4的稳定结构。最后在冠醚环内增加Li+后进行优化,得到冠醚-Li+复合物的稳定结构。
冠醚-Li+复合物中的O—Li+配位键电子密度AIM[29]拓扑分析使用了Multiwfn程序[30]。
冠醚与Li+之间的相互作用能(ΔEint)基于式(1)计算:

式中,E(tot)代表冠醚-Li+络合物的电子能量,E(crodis)和E(Li+)分别代表形变后的冠醚和Li+的电子能量。在计算E(crodis)和E(Li+)时,冠醚和Li+的位置保持与复合物中一致。ΔEint计算在B3LYPD3(BJ)/Def2QZVP级别进行,其中,对所有原子采用了更大的Def2QZVP[27-28]基函数以提高计算精度,并通过Counterpoise方法[31]考虑了基组重叠误差(BSSE)。
冠醚的几何形变能(ΔEgeom)基于式(2)计算:
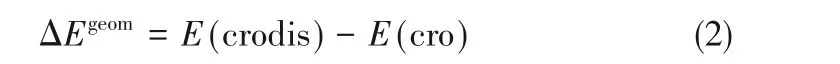
式中,E(cro)代表自由的冠醚的电子能量。
采用了自然布居分析(NPA)[32]和Mulliken[33-35]两种电荷划分方式计算原子电荷。
利用Multiwfn程序获得了电子密度差和形变后的两种冠醚分子范德华表面(isodensity=0.001 a.u.)静电势分布的空间格点,并利用VMD软件[36]绘制了电子密度差图(isovalue=0.006 a.u.)和静电势着色图。电子密度差(Δρ)基于式(3)计算:
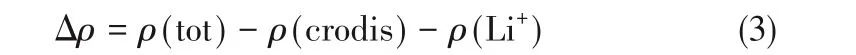
式中,ρ(tot)、ρ(crodis)和ρ(Li+)分别代表络合物、形变后的冠醚和Li+的电子密度。
基于Morokuma-Ziegler方案[37-39]的能量分解分析使用了ADF程序[40],分别在BLYP-D3(BJ)/QZ4P[23-24,41]和B3LYP-D3(BJ)/TZP两种理论级别下进行。该级别得到的ΔEint均与B3LYP-D3(BJ)/Def2QZVP级别结果符合很好。在该方案中,将ΔEint分解为式(4):
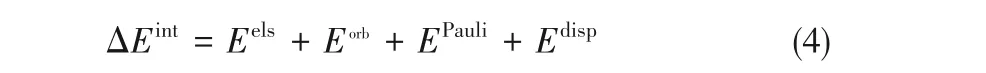
式(4)右边四项分别代表静电相互作用、轨道相互作用、Pauli排斥作用和色散作用。其中,Eels描述冠醚-Li+络合物中,冠醚与Li+的电子密度互不影响的情况下,相互之间的静电作用;Eorb通常用来描述轨道极化产生的共价相互作用,亦适用于描述本文中的O—Li+配位共价作用;EPauli用于描述冠醚与Li+之间,由于占据态轨道间Pauli排斥作用导致的络合物整体能量升高;Edisp用来描述冠醚与Li+之间的色散相互作用。
溶剂条件下的络合反应Gibbs自由能变使用Gaussian软件基于式(5)~式(7)计算:
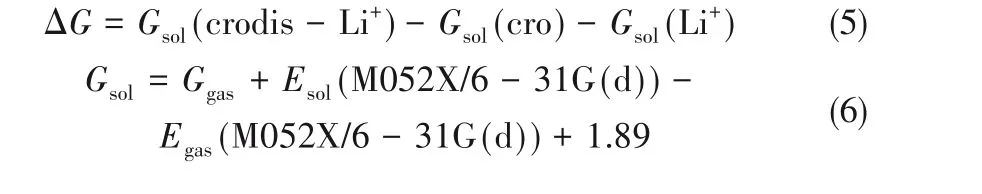

式中,Gsol、Esol和Ggas、Egas分别表示相应物质在溶剂条件下的Gibbs自由能、电子能量和在气相条件下的Gibbs自由能、电子能量,Gcorr表示Gibbs自由能的热力学矫正量,通过气相条件下的频率计算得到。Esol的计算在M052X/6-31G(d)[42-44]级别结合SMD溶剂[45]模型进行。在Gibbs自由能变计算中,所有物理量的单位统一为kcal/mol(1 kcal=4.18 kJ)。
2 结果与讨论
图2展示了5种自由冠醚以及它们络合Li+之后的优化几何结构。5个体系的详细几何参数展示在表1中,其中C—C、C—O、Si—O、Si—C和Si—Si分别表示相应化学键键长的平均值,C—Li+和Si—Li+为复合物中C和Si与Li+之间距离的平均值,On—Li+(n=1,2,3,4)分别为络合物中相应O与Li+形成的配位键键长,O的编号与图2相对应。表1中最后一行还给出了O—Li+配位键长的平均值。结合图2和表1可知,5种冠醚中的C—C、C—O键长都十分接近。冠醚与Li+结合使C—C及Si—Si键长略有增加,而C—O和Si—O键长增量更为明显。复合物中的C—Li+、Si—Li+较大(分别在2.75Å和3.1Å左右),而O—Li+较小(在1.9Å左右)。分子结构图中可以看出O与Li+有明显的相互靠近趋势。以上一系列结构特点表明Li+主要和O发生相互作用。优化所得的5种冠醚-Li+复合物均为Li+四配位模式,其中L、LSi、LSi-O和LSi-M与Li+为O—Li+四配位结构已经在晶体结构解析中得到了证实[16-17,46],而LSi/C-Li+晶体目前还未见相关报道。与Li+相互作用使冠醚的环结构发生了微弱的伸展,但是Si—C键长有所缩短,表明由于冠醚的形变及与Li+的相互作用,O与Si—O反键轨道的超共轭作用被削弱。

图2 DFT优化的稳定分子几何构象(灰色、红色、白色、青色和紫色球体分别代表C、O、H、Si和Li原子)Fig.2 DFToptimized geometric structures
通过表1可以看出,在L-Li+中,4个O—Li+配位键键长几乎相等(1.87~1.89Å),这是由于L中4个O的化学环境本质上是等价的。在LSi/C-Li+中,O1与一个Si相邻,而O1—Li+的数值较小(约1.72Å),与之处于对位的O3—Li+数值较大(约1.91Å)。经验上讲,配位键长通常与相互作用强度负相关。因此,可以认为LSi/C-Li+中的O1受Si的影响,与Li+之间产生了比另外三个O更强的相互作用。LSi-Li+中,由于O1和O4靠近Si,O2和O3远离Si,这两类O与Li的配位键长有差异。其中,靠近Si的O1/O4—Li+的键长(约1.89Å)比远离Si的O2/O3—Li+的键长(约1.94 Å)更短。Si使相邻的O—Li+配位键长更短这一现象与LSi/C-Li+中观察到的一致。但是,与L相比,—SiMe2—SiMe2—基团的引入使得LSi-Li+中的O—Li+平均配位键长增大了约0.04Å(1.88Åvs1.92Å)。继续增加掺杂Si数量到4,由于冠醚的尺寸增加,O—Li+平均配位键长被进一步拉长,并且LSi-O-Li+与LSi-M-Li+两种情况下O—Li+配位键长存在差异(1.99 Åvs1.96Å)。在LSi-O-Li+中,与1个Si相邻的O4—Li+(约1.96Å)配位键长最短,其次是与1个Si相邻的O2—Li+(约1.98Å)和与2个Si相邻的O1—Li+(1.99 Å)。在LSi-M-Li+中,4个O—Li+配位键长相等(约1.96Å),这是由于4个O的化学环境本质上是等价的。
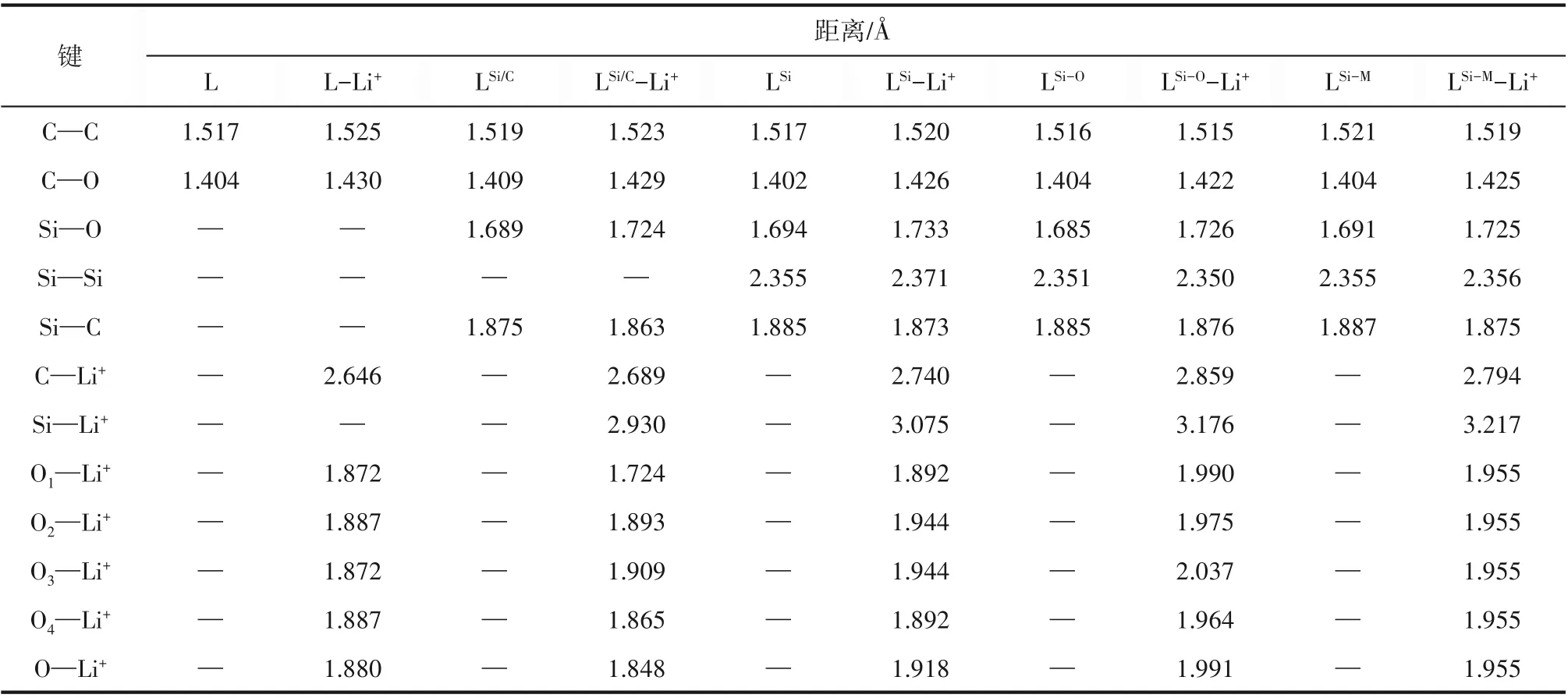
表1 自由冠醚及相应的冠醚-Li+络合物的结构参数Table 1 Geometrical parameters of the free crown ethers and the crown-Li+complexes
O—Li+配位键的电子密度AIM拓扑分析用于揭示成键本质。在每一个O—Li+配位键键径上都找到了键临界点(BCP),并分析了该位置上的电子密度(ρ)、电子密度的拉普拉斯函数(∇2ρ)、电子能量密度(H)和电子势能密度绝对值(|V|)与动能密度(G)的比值(|V|/G),相关数据展示在表2中。对于共价作用,BCP处通常∇2ρ<0、H<0、|V|/G>2,且ρ>0.14 a.u.;对于闭壳层作用(非共价作用),BCP处通常∇2ρ>0、H>0、|V|/G<1,且ρ<0.14 a.u.[47-49]。通过表2中的数据可以看出,所有O-Li+的BCP处电子密度都很小(约0.03 a.u.),且∇2ρ>0、H>0、|V|/G<1,具有明显的非共价作用特征。因此,5种冠醚与Li+形成的O—Li+配位键本质上是闭壳层相互作用。

表2 O—Li+配位键BCP处电子密度拓扑性质Table 2 Topological electron density properties at critical points of the O—Li+
为了定量地比较5种冠醚对Li+的结合能力,在3种高精度级别下计算了冠醚-Li+的相互作用能,如表3所示。结果显示,随着掺杂Si原子数量的增加,冠醚与Li+的相互作用逐步增强,且掺杂Si的相对位置也会对冠醚与Li+的相互作用产生影响。LSi/C-Li+、LSi-Li+、LSi-O-Li+、LSi-M-Li+相互作用能分别强于L-Li+约8、14、20和24 kcal/mol,表明以—SiMe2—SiMe2—单元取代—CH2—CH2—单元确实可以显著提升Si杂冠醚对Li+的络合能力,与Hänisch等[16]的研究结论一致,但与LSi、LSi-O、LSi-M三种Si杂冠醚中更长的O—Li+配位键长相悖。另外,LSi/C-Li+相互作用比LSi-Li+更强,说明在不改变冠醚大环原子数的情况下掺杂单个Si也能够提升冠醚对Li+的络合能力。考虑到冠醚的形变需要消耗能量,计算了L、LSi/C、LSi、LSi-O和LSi-M的形变能,分别为11.26、16.72、19.28、23.73和23.57 kcal/mol。随着Si掺杂数量越来越多而越来越大的形变能一定程度上削弱了Si杂冠醚对Li+的络合能力,但即便将形变能扣除,LSi/C、LSi、LSi-O和LSi-M较L仍有约2、6、7和12 kcal/mol的优势。
为了深入理解5种冠醚与Li+相互作用差异的来源,在BLYP-D3(BJ)/QZ4P和B3LYP-D3(BJ)/TZP级别计算了总相互作用能并进行能量分解分析,如表3所示。对表3纵向比较可知,5种冠醚与Li+的相互作用中静电相互作用对总的相互作用能贡献最大,轨道相互作用次之,Pauli互斥作用(正值)对相互作用起反贡献,色散作用占比较低。通过对表3横向比较,LSi/C-Li+、LSi-Li+、LSi-O-Li+和LSi-M-Li+的静电相互作用分别比L-Li+高4~5、6~7、4~6和10~11 kcal/mol,表明Si掺杂显著提升了冠醚-Li+的静电相互作用,且提升效果受到掺杂Si的相对位置的影响;LSi/C-Li+、LSi-Li+和LSi-M-Li+的轨道相互作用与LLi+相差不到1 kcal/mol,LSi-O-Li+的轨道相互作用比L-Li+弱2~4 kcal/mol,表明几何结构上的O—Li+键的拉长会略微削弱配位共价作用;对于不利于络合的Pauli互斥作用,LSi/C-Li+、LSi-Li+、LSi-O-Li+和LSi-M-Li+分别比L-Li+小1~2、6~7、15~16和12~13 kcal/mol,这主要由于总体上掺杂Si使复合物中Li+距离Si杂冠醚更远,也是Si杂冠醚对Li+络合能力更强的另一个主导因素;相比于L-Li+,Si杂冠醚-Li+中的色散相互作用也体现出一定优势(1~3 kcal/mol),这是由于Si元素的引入导致的。

表3 冠醚-Li+相互作用能及能量分解Table 3 Crown-Li+interaction energies and energy decompositions
综上分析,Si杂原子对冠醚与Li+之间静电相互作用的增强作用和对Pauli互斥的削弱作用联合主导了5种冠醚与Li+的相互作用差异,另外色散相互作用也有一定的贡献。其中,静电作用显著增强的机理仍有待进一步研究。
进一步,分析了5个冠醚-Li+体系中冠醚与Li+之间的电子密度差,以及5种形变后的冠醚分子的范德华表面静电势分布,如图3所示。这两种基于体系基态电子密度波函数的方法可以真实并直观地反映轨道相互作用和静电相互作用。
从图3的电子密度差图中可见,在5种冠醚-Li+体系中,均有显著的由O向Li+的电子密度极化(绿色),符合O—Li+配位键的化学本质。对比发现,除了LSi-O-Li+体系中O3—Li+键径上绿色等值面包裹的区域明显较小,其他4个体系中O的电子密度向Li+极化的程度十分相近。这也表明了,L-Li+、LSi/C-Li+、LSi-Li+和LSi-M-Li+4个体系中的轨道相互作用强度相近,而LSi-O-Li+体系中的轨道相互作用被较长的O—Li+配位键削弱,与前文能量分解的数据相符。
从图3的电子密度范德华表面的静电势分布图中可见,5种形变后的冠醚分子范德华表面的负静电势(红色区域)主要分布于冠醚环的4个O原子附近。形变后的L、LSi/C、LSi、LSi-O和LSi-M的表面静电势极小值分别是-69.98、-76.50、-81.27、-89.25和-91.36 kcal/mol,由此可见Si的引入显著诱导了O产生更负的静电势。进一步将两种形变的冠醚O原子对范德华表面负静电势的贡献划分在以5 kcal/mol的连续区间内,并对每个区间求面积。可见,5种冠醚的所有O原子对负静电势的总贡献(总面积)是相当的,但是随着掺杂Si的数量增加,静电势的分布越来越偏向于静电势绝对值更大的区域,因此掺杂Si使静电相互作用显著增强。另外,结合能量分解的数据可知,实际上掺杂4个Si的LSi-O与Li+的静电相互作用强度与掺杂1个Si的LSi/C相近,且弱于同样掺杂4个Si的LSi-M约6 kcal/mol。这一现象在静电势极小值及O对静电势的贡献情况中未得到充分体现。通过结构参数中的Si—Li+一项可知,与LSi-M相比,LSi-O中的Li+与Si的距离更小。因此可以推测,一方面,LSi-O中只有3个O和Si相邻,使得Si对负静电势的增强作用没能得到充分凸显;另一方面,LSi-O-Li+中Si和Li+的距离较小,使得Si—Li+静电互斥作用在更大程度上抵消了O对Li+的静电吸引作用。

图3 电子密度差图和电子密度范德华表面(isodensity=0.001 a.u.)的静电势分布图。左:电子密度差图(isovalue=0.006 a.u.),绿色和蓝色区域分别代表电子密度的积聚和衰减;中:形变后的冠醚分子的电子密度范德华表面的静电势分布图;右:O对分子的电子密度范德华表面的静电势分布的贡献Fig.3 Electron density difference diagramsand electrostatic potential distributions on the electron density van der Waals surface(isodensity=0.001 a.u.)
进而,通过表4的电荷布居分析可知,Si杂原子的引入使Si杂冠醚-Li+中的4个O原子所带的负电荷总量明显大于L-Li+,而这些负电荷增量更主要来自于靠近Si原子的O。由此可见,Si杂冠醚中的O从电负性低的Si得到了更多的电子,因此在Si杂冠醚的环中产生了更低的静电势,从而能够与Li+产生更强的静电吸引。而LSi-M-Li+中4个O所带的负电荷总量大于LSi-OLi+,说明掺杂Si的数量相同时,冠醚中与Si相邻的O数量越多,从Si得到的负电荷总量就越大。
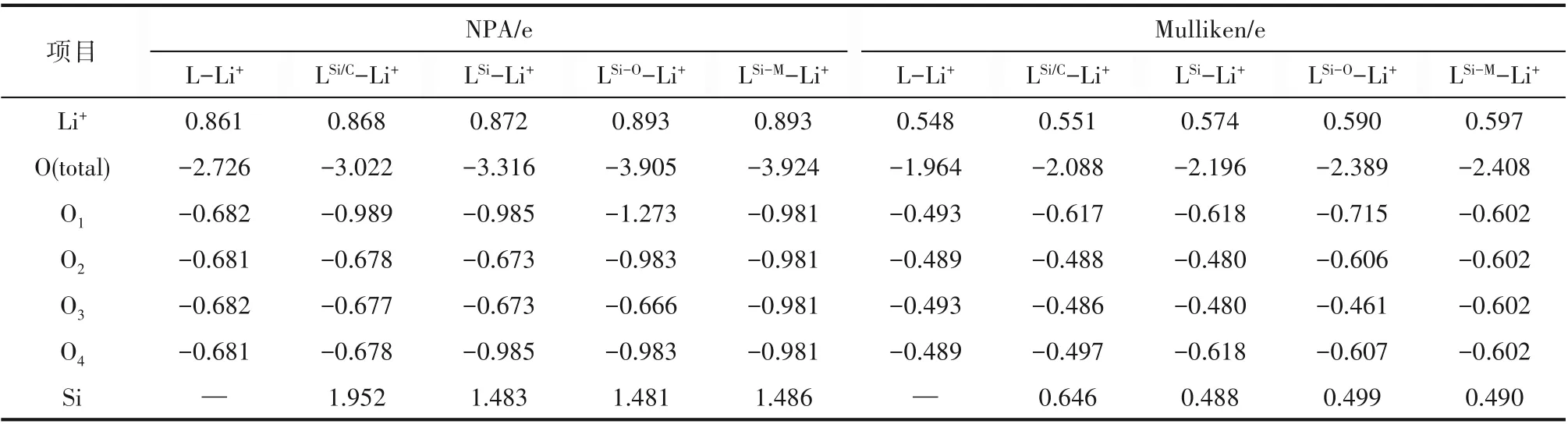
表4 冠醚-锂离子络合物中的电荷布居Table 4 Population analysis of crown-Li+complexes
Cameron等[15]的研究表明,由于Si转移大量电子给了O,带正电荷的Si与Li+之间具有库仑排斥,因此Si杂冠醚对金属离子的静电相互作用可能比原始有机冠醚更弱。但在本文所考虑的以—SiMe2—SiMe2—单 元 和—CH2—SiMe2—替 换—CH2—CH2—的Si杂12-冠-4与Li+的络合物中,Si能够自发地远离金属Li+,从而有效避免了Si-Li+库仑排斥,使得O原子的静电优势得以充分地凸显。
冠醚通常用于离子的分离、检测和反应催化,因此,冠醚-离子络合反应一般在溶剂环境下进行。计算了5种冠醚在气相条件下和二氯甲烷(DCM)溶剂条件下与Li+发生络合反应的Gibbs自由能变,结果展示在图4中。在气相条件下LSi/C、LSi、LSi-O和LSi-M与Li+的络合反应自由能变较L分别有约6、9、11和18 kcal/mol的优势。而在DCM中,LSi/C、LSi、LSi-O和LSi-M与Li+的络合反应自由能变较L分别有约3、5、6和11 kcal/mol的优势。该趋势与气相条件下相互作用能的趋势一致,说明掺杂Si原子所加强的冠醚-Li+相互作用将对溶液中冠醚-Li+络合反应产生贡献。此外,从Gibbs自由能变来看,DCM溶剂条件下的冠醚-Li+络合反应比气相条件下弱70~78 kcal/mol。这主要是由于Li+会与溶剂分子形成团簇,而冠醚络合Li+导致一部分溶剂分子被脱除,这个过程会消耗能量。

图4 冠醚-Li+络合反应的Gibbs自由能变Fig.4 Gibbs free energy changes of crown ether-Li+complexation reactions
3 结 论
本文基于量子化学密度泛函理论计算探究了12-冠-4(L)、1,1-2甲基-1-硅杂12-冠-4(LSi/C)、1,1,2,2-4甲基-1,2-2硅杂12-冠-4(LSi)、1,1,2,2,4,4,5,5-8甲基-1,2,4,5-4硅杂12-冠-4(LSi-O)和1,1,2,2,7,7,8,8-8甲基-1,2,7,8-4硅杂12-冠-4(LSi-M)与Li+的相互作用。结果表明,虽然LSi-Li+、LSi-O-Li+和LSi-M-Li+3种Si杂冠醚-Li+复合物比L-Li+具有更长的O—Li+配位键长,但是本工作中所研究的4种Si杂冠醚均具有对Li+更强的结合能力。根据结构参数、电子密度AIM拓扑分析、能量分解、电子密度差、静电势和电荷布居等一系列分析得出,O—Li+配位键具有闭壳层相互作用的特征。在LSi-Li+、LSi-O-Li+和LSi-M-Li+中,更长的O—Li+配位键长实际上仅略微削弱了配位键。Si原子由于电负性更弱而向O转移了更多电子使O带有更多负电荷,同时在络合物中Si能够自发地远离Li+,从而有效地避免了Si—Li+库仑排斥,降低了Si杂冠醚与Li+之间的Pauli排斥,充分凸显了Si杂冠醚中O的静电作用的优势。当掺杂了2组—SiMe2—SiMe2—时,LSi-M与Li+的相互作用比LSi-O更强,说明使更多O与Si相邻能令O得到更多负电荷。此外,Si的引入也使得冠醚与Li+之间的色散作用有一定程度增强。二氯甲烷中冠醚与Li+络合反应的Gibbs自由能变说明,由掺杂Si带来的相互作用能增强Si冠醚在二氯甲烷溶剂条件下络合Li+的能力。因此,为了得到与金属离子相互作用更强的Si杂冠醚,在分子合成中需要遵循以下3个规则:一是掺杂Si时不改变冠醚大环原子数;二是保证冠醚空穴和目标离子的尺寸相匹配;三是使尽量多的O与Si相邻。
本工作揭示了Si杂原子提升冠醚对Li+络合能力的根本机理,理清了一直以来对Si掺杂方式调控冠醚络合金属能力的困惑,对未来设计与合成具有高效萃取能力及其他特定功能的杂原子冠醚提供了必要的理论基础。
符号说明
Edisp,Eels,Eorb,
Epauli——分别为色散相互作用能、静电相互作用能、轨道相互作用能和Pauli互斥作用能,kcal/molE(tot),E(crodis),
E(Li+)——分别为冠醚-Li+络合物的电子能量、形变后的冠醚和Li+的电子能量,kcal/mol
Egas,Esol——分别为相应结构在气相条件下和溶剂环境中的电子能量,kcal/mol
ΔEdis,ΔEint——分别为形变能、相互作用能,kcal/mol
ESP——电子密度范德华表面的静电势,kcal/mol
G——电子的动能密度,a.u.
Gcorr——Gibbs自由能的热力学校正值,kcal/mol
Ggas,Gsol——分别为相应结构在气相条件下和溶剂环境中的Gibbs自由能,kcal/mol
ΔG——络合反应Gibbs自由能变,kcal/mol
H——电子的能量密度,a.u.
S——电子密度范德华表面的面积,Å2
V——电子的势能密度,a.u.
ρ,Δρ——分别为电子密度、电子密度差,a.u.
ρ(tot),ρ(cro),
ρ(Li+)——分别为络合物电子密度、形变后冠醚的电子密度和锂离子的电子密度,a.u.
∇2ρ——电子密度的拉普拉斯函数,a.u.
